
Dr. James Manos (MD)
January 2, 2016
January 2, 2016
Tips in Hematology
Volume (6)
2nd edition (revised)
CONTENTS
PLATELET DISORDERS AT A GLANCE
HISTORY/ SIGNS & SYMPTOMS/ CLINICAL EXAMINATION
History
Bleeding diathesis – signs & symptoms
Clinical examination
Clinical evaluation of bleeding disorders
Features of generalized hemostatic defect
CBC/FBC (complete/ full blood count)
Platelet count – limitations
Peripheral blood smear
Basic coagulation studies
Biochemistry
Urinalysis
Infections
Medications
Petechia
Petechia – causes
Purpura & ecchymosis
Purpura & ecchymosis – causes
Blood in stool
Bleeding gingiva (gums)
Prolonged epistaxis (nosebleed)
Soft tissue bleeding
Joint damage/ arthritis
Retinal bleeding
Severe hemorrhage/ exsanguination
Anemia
Cerebral hemorrhage
DEFECTS OF HEMOSTASIS
Defects of hemostasis
Defects in primary hemostasis
Defects in secondary hemostasis
LABORATORY PARAMETERS OF COAGULATION
Bleeding time (BT)
Causes of prolonged bleeding time
Mean platelet volume (MPV) & platelet distribution width (PDW)
High MPV - causes
Abnormally low MPV – causes
PFA-100
Causes of prolongation of both test results (Col/Epi >180 seconds, Col/ADP >120 seconds)
Ristocetin-induced platelet aggregation (RIPA)
Prothrombin time (PT) and INR
Parameters that affect PT
Prolonged PT – causes
Activated partial thromboplastin time (APTT)
Prolonged APTT – causes
Thrombin time (ΤΤ)
Prolonged TT – causes
Interpretation of the screening test of hemostasis in a bleeding disorder
DIC (disseminated intravascular coagulation)
THROMBOCYTOSIS & METABOLIC EFFECTS
PLATELET DISORDERS AT A GLANCE
HISTORY/ SIGNS & SYMPTOMS/ CLINICAL EXAMINATION
· History: Fever/ sepsis? Drugs (see below). Transfusion (see below). Family history.
· Bleeding diathesis – signs & symptoms: petechia/ purpura/ ecchymosis, easy bruising, gingival bleeding (bleeding gums), easy bruising, menorrhagia (heavy & prolonged menstrual periods), epistaxis, abnormally prolonged bleeding from small injuries, postpartum bleeding, gastrointestinal (GI) bleeding, hemarthrosis, perioperative and postoperative bleeding, and bleeding after dental or minor surgical procedures.
· Clinical examination: Hepatosplenomegaly? Hypersplenism? Lymphadenopathy? Petechia/ purpura/ ecchymosis.
· Clinical evaluation of bleeding disorders: deficient hemostasis due to an abnormality of vascular walls, platelets, or coagulation factors. Clinical evaluation: Is the bleeding suggestive of a primary or secondary hemostatic defect? Is the bleeding disorder inherited or acquired? Is the nature of bleeding indicative of a primary or secondary hemostatic disorder?
· Features of generalized hemostatic defect: easy, spontaneous bleeding, excessive bleeding relative to the degree of trauma, repeated episodes of bleeding, multiple sites of bleeding, and similar family history in a close relative.
· CBC/FBC (complete/ full blood count): Platelets (also MPV)? WBCs? RBCs? Reticulocytes increased?
· Platelet count - limitations: megakaryocytes, giant platelets, clumped platelets, or platelet satellitism may falsely decrease the platelet count. Fibrin clots may affect the platelet count.
· Elevated platelet levels may occur after splenectomy.
· A platelet count may be performed on samples collected from sodium citrate anticoagulant tubes (light blue top) when EDTA (purple top) results exhibit platelet clumping or platelet satellites.
· Peripheral blood smear: platelet size (giant platelets?); agglutination of platelets?
· Basic coagulation studies: PT/ INR, APTT, +_ bleeding time.
· Biochemistry: LFTs (liver function tests (enzymes)), BUN/ Urea, Creatinine, LDH.
· Urinalysis: hematuria? Proteinuria?
· Viral infections: EBV (e.g., Monospot), CMV, HBV, HCV, HIV?
· Medications: many; e.g., cimetidine, ranitidine, heparin, TMP (trimethoprim), penicillin, furosemide, vancomycin, teicoplanin, anticoagulants/ antiplatelet (blood thinners), corticosteroids, NSAIDs (nonsteroidal anti-inflammatory drugs), etc.
· Petechia: pin point <_3 mm.
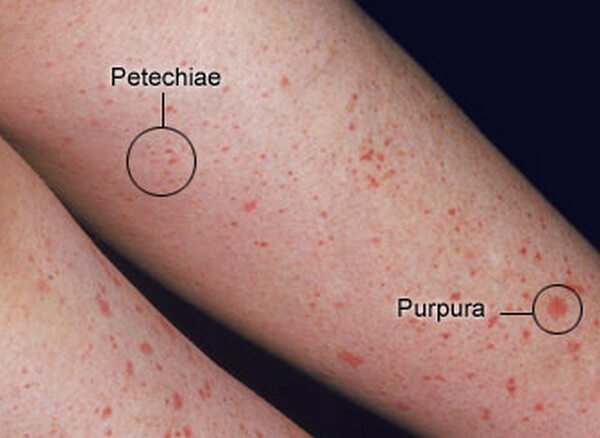{kind=link}
· Petechia – causes, e.g., leukemia (acute & chronic), vitamin K deficiency, Wiskott – Aldrich syndrome. For an extended catalog of causes, see https://en.wikipedia.org/wiki/Petechia#Causes
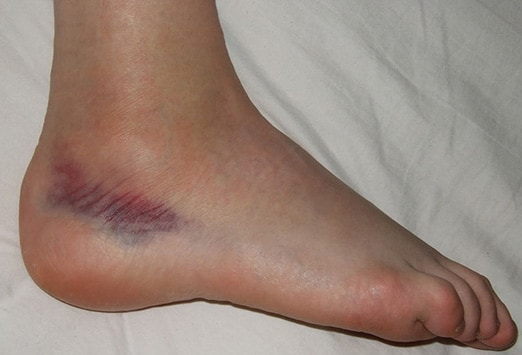
· Purpura & ecchymosis: purpura: >3mm, < 1 cm; ecchymosis: >1 cm.
· Purpura:
{kind=link}
· Ecchymosis:
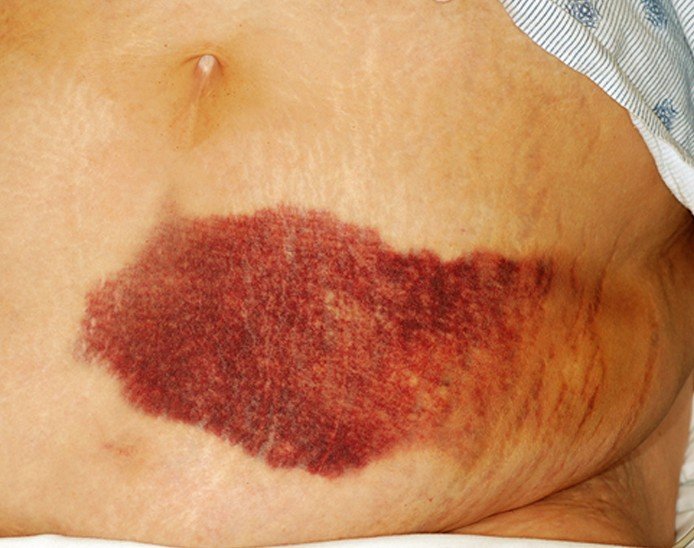{kind=link}
{kind=link}
{kind=link}
· Purpura & ecchymosis – causes: e.g., leukemia (acute & chronic), vitamin K deficiency.
· Causes of purpura: thrombocytopenic purpura (primary or secondary), post-transfusion purpura, hypertensive states, senile purpura (easy damage of small vessels), vasculitis (e.g., purpura Henoch – Schönlein; especially in children; arthritis, abdominal pain, and purpura – especially in legs & buttocks), DIC, scurvy, cocaine + levamisole (veterinary deworming agent), infections (rickettsia; meningitis).
· Causes of ecchymosis: There are many causes. Coagulopathies, including hemophilia, may cause ecchymosis in children.
· Blood in stool: e.g., acute leukemia, Wiskott – Aldrich syndrome (especially in infancy).
· Bleeding gingiva (gums): e.g., leukemia (acute & chronic), Wiskott – Aldrich syndrome.
· Prolonged epistaxis (nosebleed): e.g., Wiskott – Aldrich syndrome.
· Soft-tissue bleeding: e.g., hemophilia, von Willebrand’s disease.
· Joint damage/ arthritis: e.g., hemophilia, von Willebrand’s disease.
· Retinal bleeding: e.g., acute leukemia.
· Severe hemorrhage/ exsanguination: e.g., von Willebrand’s disease, acute leukemia, vitamin K deficiency.
· Anemia: e.g., von Willebrand’s disease.
· Cerebral hemorrhage: e.g., Wiskott – Aldrich syndrome.
DEFECTS OF HEMOSTASIS
· Defects of hemostasis:
· a) Inherited: presentation in early childhood; +_ family history; a certain inheritance pattern present, e.g., autosomal dominant, recessive, or X-linked.
· b) Acquired: occurs at any age; no family history; underlying disease or predisposing factor present.
· Defects in primary hemostasis: petechiae, purpura, bleeding gums, epistaxis, menorrhagia; no delayed bleeding.
· Lab investigations: bleeding time, platelet count & blood smear.
· Specific tests: bone marrow examination, platelet function studies.
· Defect in secondary hemostasis: deep hematoma, hemarthrosis, delayed bleeding. Lab investigations: PT, aPTT, TT (thrombin time), mixing studies, coagulation factor assays, FDPs (fibrinogen/fibrin degradation products).
LABORATORY PARAMETERS OF COAGULATION
· Bleeding time (BT): assesses the platelets’ function. It involves making a patient bleed and then timing how long it takes for them to stop bleeding following a skin puncture/incision. Both the Ivy and the Duke methods require a sphygmomanometer.
· In the Ivy method, the blood pressure cuff is placed on the upper arm and inflated to 40 mmHg. A lancet or scalpel blade is used to make a shallow incision that is 1 millimeter deep on the underside of the forearm. A standard-sized incision is made around 10 mm long and 1 mm deep. The time from incision to when all bleeding has stopped is measured and called the bleeding time. Every 30 seconds, a filter paper or a paper towel is used to draw off the blood. The test is finished when the bleeding has stopped completely. A normal value is less than 9.5 minutes.
· With the Duke method, the patient is pricked with a special needle or lancet, preferably on the earlobe, after having been swabbed with alcohol. The prick is about 3 – 4 mm deep. The patient then wipes the blood away every 30 seconds with a piece of filter paper. The test ceases when bleeding ceases. The usual time is about 2–5 minutes.
· Causes of prolonged bleeding time: thrombocytopenia, disorders of platelet function, vWD (von Willebrand’s disease), and vascular disorders. Specifically, bleeding time is prolonged in DIC (disseminated intravascular coagulation), vWD (von Willebrand’s disease), thrombocytopenia, drugs (aspirin), end-stage liver failure, uremia, congenital afibrinogenemia, Glanzmann’s thrombasthenia, and Bernard–Soulier syndrome.
· Mean platelet volume (MPV) & platelet distribution width (PDW): Mean platelet volume (MPV) and platelet distribution width (PDW) are calculations performed by automated blood analyzers.
· MPV reflects the average size of platelets present in a person's sample of blood.
· PDW indicates how uniform the platelets are in size.
· These calculations can give information about platelets and/or about the cause of a high or low platelet count. Larger platelets are usually relatively young and more recently released from the bone marrow, while smaller platelets may be older and have been in circulation for a few days. PDW measures the range of platelet sizes in a blood sample. This gives an idea of the amount of active platelet release. According to one study, MPV and PDW are simple platelet indices that increase during platelet activation. PDW is a more specific marker of platelet activation since it does not increase during simple platelet swelling.
· MPV is a machine-calculated measurement of the average size of platelets in the peripheral blood and is included in a CBC (complete blood count). The normal range of MPV is 7.5–11.5 fL. Since the average platelet size is larger when the body produces more platelets, the MPV test results can be used to infer platelet production in the bone marrow or platelet destruction.
· High MPV – causes: A high number of large platelets (high MPV) in a person with a low platelet count suggests the bone marrow is producing platelets and releasing them into circulation rapidly. MPV is elevated when platelet destruction occurs, and this may be seen in inflammatory bowel disease (IBD), immune thrombocytopenic purpura (ITP), myeloproliferative disorders, and Bernard–Soulier syndrome. It may also occur in pre-eclampsia and recovery from transient hypoplasia.
· Abnormally low MPV – causes: Abnormally low MPV values correlate primarily with thrombocytopenia when it is due to impaired production, such as in aplastic anemia.
· PFA-100: a platelet function analyzer that aspirates blood in vitro from a blood specimen into disposable test cartridges through a microscopic aperture cut into a biologically active membrane at the end of a capillary. The membrane of the cartridges is coated with collagen and either adenosine diphosphate (ADP) or epinephrine, thereby inducing platelet plug formation that closes the aperture. The PFA test result depends on platelet function, plasma von Willebrand Factor level, platelet count, and (to some extent) hematocrit. The PFA test is initially performed using the Collagen/Epinephrine membrane. A normal Col/Epi closure time (<180 seconds) excludes the presence of a significant platelet function defect. If the Col/Epi closure time is prolonged (>180 seconds), the Col/ADP test is automatically performed. Aspirin-induced platelet dysfunction is most likely if the Col/ADP result is normal (<120 seconds).
· Causes of prolongation of both test results (Col/Epi >180 seconds, Col/ADP >120 seconds): anemia (hematocrit <28%); thrombocytopenia (platelet count < 100 x 109/L); a significant platelet function defect other than aspirin, including von Willebrand disease and inherited/acquired platelet dysfunction.
· Ristocetin-induced platelet aggregation (RIPA): an ex vivo assay for live platelet function. It measures platelet aggregation using von Willebrand factor (vWF) and the exogenous antibiotic ristocetin, added in a graded fashion.
· It is used to diagnose von Willebrand’s disease (vWD) and Bernard–Soulier syndrome. In an unknown manner, the antibiotic ristocetin causes von Willebrand factor to bind the platelet receptor glycoprotein Ib (GpIb), so when ristocetin is added to normal blood, it causes agglutination of fixed platelets or initiates the initial agglutination phase of aggregation of live platelets. In von Willebrand disease, where von Willebrand factor is absent or defective, abnormal agglutination occurs.
· Hemostasis – Coagulation Cascade
· See:
{kind=link}
{kind=link}
· Prothrombin time (PT) and INR: tissue thromboplastin & calcium (Ca++) are added to platelet-poor plasma, and the clotting time of the mixture is noted. The prothrombin time (PT), along with the international normalized ratio (INR), is an assay used to evaluate the extrinsic and common pathways of coagulation. The result of a prothrombin time performed on a normal individual will vary depending on the type of analytical system employed, due to differences among manufacturers' tissue factor used in the reagent and across batches. The INR was devised to standardize the results. The INR is the ratio of a patient's prothrombin time to that of a normal (control) sample, raised to the power of the ISI (International Sensitivity Index) value for the analytical system used. The equation is INR = (PT test/PT normal) raised to the ISI exponent. PT normal is defined as the geometric mean of the prothrombin times (PT) of a reference sample group. PT/INR is used to determine the clotting tendency of blood, measure the effects of warfarin dosage, assess liver damage, check for vitamin K deficiency, and monitor the vitamin K effect on lowering PR/ INR when used for warfarin overdose treatment. Procedure: blood is drawn into a test tube containing liquid sodium citrate, which acts as an anticoagulant by binding the calcium in a sample. For accurate measurement, the blood-to-citrate ratio must be fixed and labeled on the side of the measuring test tube by the manufacturer. The blood is mixed and then centrifuged to separate blood cells from plasma (in newborns, a capillary whole-blood specimen is used). A plasma sample is extracted from the test tube and placed into a measuring test tube. Following this, an excess of calcium is added to a phospholipid suspension in the test tube, thereby reversing the effects of citrate and enabling the blood to clot again. Eventually, to activate the extrinsic/tissue factor clotting cascade pathway, tissue factor (AKA factor III) is added, and the time it takes for the sample to clot is measured optically. Some laboratories use a mechanical measurement, which eliminates interferences from lipemic and icteric samples.
· PT is a measure of extrinsic and common pathways.
· Each laboratory should establish its own normal range, but in general, the prothrombin time (PT) for a normal plasma sample in the reference range is between 13 and 15 seconds.
· The International Normalized Ratio (INR) is the PT ratio of a test sample compared to a normal PT (derived from the log mean normal prothrombin time (LMNPT) of 20 normal donors) corrected for the sensitivity of the thromboplastin used in the test. It is the Prothrombin Time Ratio value obtained using the first WHO Reference Thromboplastin with an ISI of 1.0.
· INR = [PT patient / PT reference plasma]ISI or INR = [PT ratio]ISE (ISI is the international sensitivity index), e.g., for a patient on warfarin with a PT of 23 seconds and a mean normal PT of 12 seconds using a thromboplastin with an ISI of 1.2, the INR is 2.18.
· The INR is typically used to monitor patients on warfarin or related oral anticoagulant therapy. The normal range for a healthy person not using warfarin is 0.8 – 1.2, and for people on warfarin therapy, an INR of 2.0–3.0 is usually targeted (note from the writer of this text: although on the very elderly patients, high INR towards the upper limit of 3 may be correlated with increased bleeding risk), although the target INR may be higher in particular situations, such as for those with a mechanical heart valve. In patients with prosthetic heart valves, recommendations vary as to the target INR. The following is offered as a general guideline, but remember that therapy must be individualized. For bioprosthetic valves, the INR target is 2–3 for 3 months following implantation; anticoagulation may then be discontinued unless the patient has another indication, such as atrial fibrillation or development of prosthetic valve thrombosis. For mechanical valves, the target INR is a) For the aortic valve, the target INR is 2 – 3. b) For the mitral valve, the target INR is 2.5 – 3.5. Patients with atrial fibrillation (AF) should be kept at the higher end of this range. In patients with low hemorrhage risk, low-dose aspirin is recommended in addition to warfarin.
· If the INR is outside the target range, a high INR indicates a higher risk of bleeding, while a low INR suggests a higher risk of developing a clot.
· Parameters that affect PT: lipemia and hyperbilirubinemia interfere with the detection of clot formation by photo-optical methods. The results of the PT may be affected by a wide variety of factors, including the manner of blood coagulation, the type of container, the type of anticoagulant, specimen transport and storage conditions, incubation time and temperature, assay reagents, and the method of endpoint detection. Many physiologic factors can also influence the PT. The PT is prolonged in cord blood and newborns due to relatively low levels of vitamin K-dependent clotting factors, which may not increase to the normal adult range until several weeks after birth. Lipemia, hyperbilirubinemia, and hemolysis interfere with the detection of clot formation by photo-optical methods and cause falsely elevated PT (prothrombin time) values. Heparin at therapeutic doses usually does not interfere with PT, but PT prolongation can lead to patients receiving higher doses of Heparin. In fact, due to variability in the sensitivities of different thromboplastins to heparin, a falsely prolonged PT can occur during the initiation of warfarin treatment when the patient is simultaneously receiving heparin therapy.
· Prolonged PT – causes vitamin K deficiency, oral anticoagulation therapy (warfarin), DIC (disseminated intravascular coagulation), inherited lack of factors in extrinsic or common pathways, liver disease, congenital afibrinogenemia, factor V deficiency, factor X deficiency (as seen in amyloid purpura).
· Activated partial thromboplastin time (aPTT or APTT): Platelet–poor plasma is incubated with an activator; then phospholipid and calcium are added, and clotting time is noted. The historical name is 'kaolin-cephalin clotting time,' using kaolin and cephalin as materials. The PTT measures the time it takes for blood to clot, using two consecutive biochemical reactions: the intrinsic and common coagulation pathways. It detects abnormalities in blood clotting and is also used to monitor the treatment effect of heparin. Procedure: blood is drawn into a test tube containing oxalate or citrate, molecules that act as anticoagulants by binding calcium in the sample. The blood is mixed and then centrifuged to separate blood cells from plasma. A plasma sample is extracted from the test tube and placed into a measuring test tube. Following this, an excess of calcium is added to a phospholipid suspension, which is then mixed into the plasma sample to reverse the anticoagulant effect of oxalate, enabling the blood to clot again. Eventually, to activate the intrinsic pathway of coagulation, an activator (such as silica, celite, kaolin, or ellagic acid) is added, and the time it takes for the sample to clot is measured optically. Some laboratories use a mechanical measurement that eliminates interference from lipemic and icteric samples.
· APTT is a measure of the intrinsic pathway (contact activation pathway) and the common path.
· The reference range for the clotting time for the APTT is between 27 and 35 seconds. However, this varies widely between laboratories and depends on factors such as whether the test is automated or manual, the type of activator, and the incubation times used.
· Prolonged aPTT – causes inherited deficiency of factors VIII (hemophilia A) and IX (hemophilia B); circulating inhibitors of coagulation (e.g., lupus anticoagulant, factor VIII inhibitor), DIC (disseminated intravascular coagulation), heparin therapy, advanced liver disease, factor V deficiency, factor X deficiency (as seen in amyloid purpura), factor XII deficiency, congenital afibrinogenemia.
· On vitamin K deficiency or oral anticoagulation (warfarin), aPTT is normal but may be mildly prolonged.
· On von Willebrand’s disease, APTT is prolonged or unaffected.
· On c1 esterase inhibitor deficiency (on hereditary angioedema), aPTT is shortened.
· Thrombin time (ΤΤ): The thrombin time compares the rate of clot formation to that of a sample of normal pooled plasma. Thrombin is added to plasma samples. In blood samples containing heparin, batroxobin (AKA reptilase; a substance derived from snake venom) is used instead of thrombin (batroxobin is not inhibited by heparin). Normal values for thrombin time are 12-14 seconds (15-20 seconds if batroxobin is used). Thrombin time can be prolonged by fibrin degradation products, heparin, and fibrinogen deficiency or abnormality.
· Each laboratory must establish its own reference range, but in general, the reference range for the thrombin time is in the region of 13-15 sec.
· Prolonged TT – causes disorders of fibrinogen (afibrinogenemia/ hypofibrinogenemia/ dysfibrinogenemia), heparin treatment, chronic liver disease, FDPs (fibrinogen/fibrin degradation products), and unfractionated heparin administration.
· Interpretation of screening test of hemostasis in a bleeding disorder:
· a) Prolonged bleeding time; normal Platelet count, PT, APTT: defect in platelet function or vascular disorders; common causes: vWD (von Willebrand’s disease), aspirin, uremia, storage pool defect, Glanzmann’s thrombasthenia.
· b) Prolonged bleeding time; decreased platelet count; normal PT and APTT: thrombocytopenia; common causes: secondary causes, drugs, ITP (idiopathic thrombocytopenic purpura).
· c) Prolonged PT; normal bleeding time, platelet count, APTT: extrinsic pathway defect; common causes: oral anticoagulants (warfarin), Vitamin K deficiency, deficiency of factor VII, liver failure – early stage.
· d) Prolonged APTT; normal bleeding time, platelet count, PT: intrinsic pathway defect; common causes: heparin, hemophilia A (factor VIII deficiency) or B (factor IX deficiency), factor XII deficiency, vWD (von Willebrand’s disease), inhibitors.
· e) Prolonged PT and APTT; normal bleeding time and platelet count: common pathway defect; common causes: heparin, liver disease, vitamin K deficiency, oral anticoagulants (warfarin), deficiency of factors V, X (as seen in amyloid purpura), II, I.
· f) Prolonged PT, APTT, bleeding time; decreased platelet count: multiple pathways defect; common causes: DIC (disseminated intravascular coagulation), liver failure – end-stage.
· g) Normal bleeding time, platelet count, PT, APTT: common causes: mild vWD (von Willebrand’s disease), vascular disorder, platelet function defect, factor XIII deficiency.
· h) Prolonged PT; normal or mildly prolonged APTT; normal bleeding time & platelet count: common causes: vitamin K deficiency or oral anticoagulation (warfarin).
· i) Prolonged bleeding time; prolonged or normal APTT; normal PT and platelet count: common causes: von Willebrand's disease.
· j) Shortened APTT; normal PT, bleeding time & platelet count: causes: c1 esterase inhibitor deficiency (hereditary angioedema).
· k) Prolonged PT, APTT & bleeding time; normal platelet count: causes: congenital afibrinogenemia.
· l) Prolonged bleeding time; decreased or normal platelet count; normal PT & APTT: causes: Bernard – Soulier syndrome.
· DIC (disseminated intravascular coagulation) is an acquired disorder that occurs in a wide spectrum of underlying conditions.
· Characteristics:
· i) Widespread systemic activation of coagulation with the formation of microthrombi (small clots) in small blood vessels.
· ii) Bleeding diathesis secondary to depletion of coagulation factors and platelets.
· Mechanism: activation of coagulation factors and platelets; formation of microthrombi in circulation; end-organ damage and depletion of coagulation factors and platelets due to their consumption, leading to bleeding diathesis.
· Causes: sepsis/ severe infections, trauma (especially brain or crush injury), obstetric problems (amniotic fluid embolism, abruption placenta, septic abortion, eclampsia, intrauterine retention of dead fetus), malignancy (disseminated solid cancer, acute promyelocytic leukemia), severe hemolytic transfusion reactions, thermal injury (heat stroke, extensive burns), snakebite (e.g., Russell’s viper), severe liver disease, giant hemangioma (Kasabach – Merritt syndrome). Also, AML (acute myeloid leukemia) subtype M3 (APL, acute promyelocytic leukemia) chemotherapy may be complicated when promyelocytes release granule contents into the peripheral circulation.
· For causes, see: http://emedicine.medscape.com/article/199627-overview#a7
· Clinical features: sudden onset of spontaneous bleeding from multiple sites (petechia, purpura, hematemesis, melena, hematuria, epistaxis, oozing from venipuncture sites); purpura fulminans (patchy areas of hemorrhagic skin necrosis) and presence of an underlying condition associated with DIC.
· Lab features: reduced platelets or falling platelets on repeat testing; increased D–dimers, increased FDPs (fibrinogen/ fibrin degradation products); prolonged PT & aPTT; reduced Fibrinogen or falling level on repeat testing. (but, as fibrinogen is an acute phase reactant, it may be normal or even increased by 57%). Low fibrinogen levels may also be measured in advanced liver diseases such as cirrhosis.
· The massive fibrin deposition in DIC suggests that fibrinogen levels would be decreased. Thus, fibrinogen measurement has been widely advocated as a useful tool for the diagnosis of DIC; however, it is not very helpful. Fibrinogen, as a positive acute-phase reactant, is increased in inflammation; although values may decrease as the illness progresses, they are rarely low. One study demonstrated that in up to 57% of DIC patients, fibrinogen levels may remain within normal limits.
· Also, low plasma levels of coagulation inhibitors (ATIII (antithrombin III) or protein C).
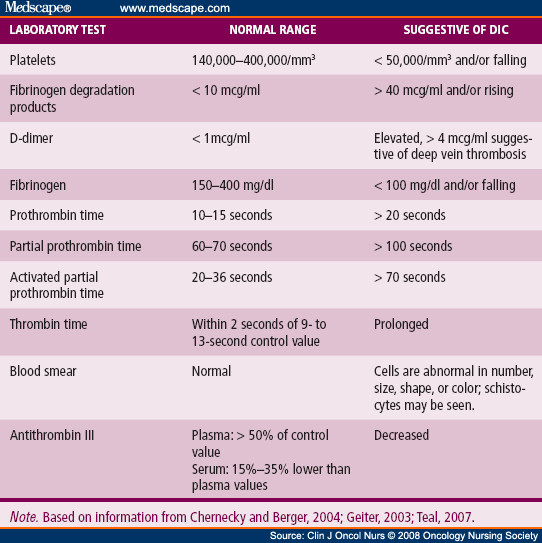{kind=link}
· Blood smear may show schistocytes (fragmented red cells), indicative of microangiopathic hemolytic anemia (MAHA) (intravascular hemolysis).
· The peripheral blood smear can reveal schistocytes, though these are rarely seen to exceed 10% of red blood cells (RBCs). The presence of schistocytes is neither sensitive nor specific for DIC, but in certain instances, it may help confirm a diagnosis of chronic DIC when schistocytes are seen alongside normal coagulation values and elevated D-dimer levels.
· Mortality of DIC: 10 – 50%.
· International Society on Thrombosis and Hemostasis (ISTH) score calculation for the laboratory diagnosis of DIC: http://www.cancernetwork.com/sites/default/files/figures_diagrams/1502FeinsteinTable.png
{kind=link}
· and
{kind=link}
· There is also the Japanese Association for Acute Medicine Scoring System for DIC.
· For both score systems, see: http://emedicine.medscape.com/article/199627-workup#c7
· A multicenter, prospective study concluded that the Japanese Association for Acute Medicine (JAAM) DIC scoring system exhibits good prognostic value in predicting multiple organ dysfunction syndromes (MODS) and poor prognosis in patients with severe sepsis, and can detect more patients requiring treatment. Conducting repeated daily JAAM scoring improves the ability to predict the patient’s prognosis (Reference: http://www.ccforum.com/content/pdf/cc12783.pdf).
· A prospective study concluded that in sepsis, the Japanese Association for Acute Medicine (JAAM) DIC score identified most of the patients diagnosed by the overt International Society on Thrombosis and Hemostasis (ISTH) criteria but failed to discriminate between survivors and non-survivors amongst DIC patients (Reference: http://www.ncbi.nlm.nih.gov/pubmed/22138415 ).
· Chronic DIC: DIC exists in both acute and chronic forms. Acute DIC develops when sudden exposure of blood to procoagulants, such as tissue factor (TF) or tissue thromboplastin, generates intravascular coagulation. Compensatory hemostatic mechanisms are quickly overwhelmed. Consequently, severe consumptive coagulopathy occurs, leading to hemorrhage development. Abnormalities of blood coagulation parameters are readily identified. Acute DIC frequently results in (multi) organ failure. In contrast, chronic DIC reflects a compensated state that develops when blood is continuously or intermittently exposed to small amounts of TF. Compensatory mechanisms in the liver and bone marrow are not overwhelmed, and there may be little obvious clinical or laboratory evidence of DIC. Chronic DIC is more frequently observed in patients with solid tumors and in those with large aortic aneurysms.
· Differential diagnosis: dysfibrinogenemia, hemolytic–uremic syndrome (HUS) & thrombotic thrombocytopenic purpura (TTP), heparin-induced thrombocytopenia (HIT), idiopathic thrombocytopenic purpura (ITP).
· Treatment: treatment of DIC is centered around treating the underlying condition ( DIC can result from numerous clinical conditions, including sepsis, trauma, obstetric emergencies, and malignancy). Transfusions of platelets or fresh-frozen plasma (FFP) can be considered in cases of significant bleeding or in those with a planned invasive procedure. Platelet and factor replacement should be directed not at simply correcting laboratory abnormalities but at addressing clinically relevant bleeding or meeting procedural needs. The target goal of such transfusion depends on the clinical situation. Cryoprecipitate can be considered for those with low fibrinogen levels. Treatment of thrombosis with anticoagulants such as heparin is rarely used due to the risk of bleeding. Heparin should be provided to those patients who demonstrate extensive fibrin deposition without evidence of substantial hemorrhage; it is usually reserved for cases of chronic DIC. Heparin is appropriate for treating thrombosis associated with DIC. It also has limited use in acute hemorrhagic DIC in a patient with a self-limited condition of acral cyanosis and digital ischemia. Antithrombin and antifibrinolytics are generally not used. Studies have shown that recombinant human activated protein C, previously recommended for those with severe sepsis and DIC, confers no benefit and is not used today. Recombinant factor VII has been proposed by some studies as a ‘last resort’ in those with severe hemorrhage due to obstetric or other causes, but conclusions about its use are still insufficient, and it may also increase thrombosis risk. With this drug, the risk of thrombosis, particularly arterial thromboembolic events (e.g., myocardial ischemia, MI, cerebral ischemia, and/or infarction), may be further increased in non-hemophilic patients who receive factor VIIa (recombinant) for non-FDA-labeled indications. A potentially greater risk occurs in patients with disseminated intravascular coagulation (DIC), advanced atherosclerotic disease, crush injuries, septicemia, or concomitant treatment with activated or nonactivated prothrombin complex concentrates (APCCs or PCCs) due to circulating tissue factor (TF) or predisposing coagulopathy.
THROMBOCYTOSIS & METABOLIC EFFECTS
· If a person's platelet count is increased, serum LDH can be artificially high and not reflect the LDH present in the circulation.
· Pseudohyperkalemia is also positively correlated to (1) thrombocytosis due to the release of potassium from platelet granules during coagulation, (2) erythrocytosis due to the dilution of the released potassium in smaller volumes of serum, and (3) the presence of activated platelets, which have the capability of aggregation at a higher speed and release more potassium during degranulation.
· A human study concluded that pseudohyperkalemia is mainly present in patients with thrombocytosis or mixed-type disorders, probably as a result of the degranulation of platelets, which offers a potassium load to the surrounding plasma at the time of clot formation in vitro. However, the degree of pseudohyperkalemia does not increase proportionally with increases in platelet counts, which may be due to the transfer of part of the potassium load from plasma back into red and white blood cells.
· Another human study concluded that the difference between serum and plasma potassium concentration (Dk) is increased in patients with erythrocytosis, thrombocytosis, or both. This phenomenon is more profound in patients with a mixed-type disorder, such as polycythemia vera patients, compared to those with erythrocytosis alone.
No comments:
Post a Comment