
Dr. James Manos (MD)
January 5, 2016
(Corrected: 26 August 2018)
(Corrected: 26 August 2018)
Review: Tips in Medical Biochemistry
Volume (1)
Volume (1)
CONTENTS
Sepsis
Markers of sepsis
ICU (intensive care unit)
APACHE II score
Endocrinology
Parathyroid disorders
Hypoparathyroidism
Pseudohypoparathyroidism
Pseudohypoparathyroidism – Laboratory investigation
Pseudopseudohypoparathyroidism
Hyperparathyroidism
Racquet nails (image)
Hyperparathyroidism – characteristics & laboratory investigation
Parathyroid hormone-related protein (PTHrP)
Vitamin D
Thyroid disorders
Thyroid function tests (TFTs)
Hyperthyroidism
Interpretation of TFTs (thyroid function tests) on hyperthyroidism
Hypothyroidism
Interpretation of TFTs (thyroid function tests) on hypothyroidism: a) Raised TSH; low FT4 or FT3
Subclinical thyroid disease
Algorithm for interpretation of thyroid function tests (TFTs)
Interpretation of thyroid function tests on patients taking thyroxine (T4)
Calcitonin
Adrenal disorders
ACTH stimulation test (Synacthen test; tetracosactide test; Cosyntropin test)
Diagnostic chart for ACTH stimulation test
Diagnostic algorithms for adrenal insufficiency
Addison’s disease
Hyperaldosteronism
Diagnostic algorithm for diagnosing primary aldosteronism
Dexamethasone suppression test (DST)
Cushing’s syndrome, Cushing’s disease, ectopic Cushing’s & pseudo – Cushing’s
Diagnostic algorithm for Cushing’s syndrome
Pheochromocytoma
Diagnostic algorithm for suspicion of pheochromocytoma
SEPSIS
· Markers of sepsis: procalcitonin (PCT; increased in sepsis), WBCs (white blood cells), CRP, Eosinopenia (cut off < 50 cells/ mm3), PT (prolonged), APTT (prolonged), prealbumin (transthyretin), IL-6, IL -8, IL -10, IL – 12 & IP – 10 (interferon induced protein) (the last 2 on paediatric newborn), IL -18 (Gram (-) sepsis), lactic acid, glycoprotein 130, IL – 1 R (receptor), IL-2 R, ferritin, LBP (lipopolysacharide binding protein), presepsin, SAA (serum amyloid alpha), soluble CD14, MMP – 9 (metaloproteinases), phospholipase A2, protein C (decreased), antithrombin (decreased), D – Dimers (increased), PAI -1 (plasminogen activator inhibitor – 1), protein S, thrombomodulin (decreased), fibrin (increased; in bacterial sepsis, especially Gram (-)), nCD64 (neutrophil CD64 receptor), ANP, BNP, endothelin – 1 & S100 calcium binding protein B (sepsis related encephalopathy), PBEF (pre B cell colony enhancing factor), NO (increased), ceruloplasmine (liver dysfunction), mannan & antimannan Abs (antibodies) (invasive fungal infections), IP – 10 (inducible protein – 10), Interferon – γ (gamma) (viral infections) etc.
ICU (intensive care unit)
· APACHE II score: severity of diseases classification system. Includes the following parameters: rectal temperature, mean arterial pressure, heart rate, respiratory rate, oxygenation A – aDO2 or FIO2, arterial PH, serum HCO3 (if no ABGs), serum sodium, serum potassium, serum creatinine, hematocrit, WBCs (white blood cells), GCS (Glasgow comma scale), age, chronic health points.
· APACHE II score: http://www.medicalcriteria.com/criteria/uti_apache2.htm
Endocrinology
Parathyroid disorders
· Hypoparathyroidism: causes hypocalcemia (low blood calcium)
· Signs & symptoms: paresthesia, tingling around the mouth, hands & feet, muscle spasms that may affect hands & feet (tetany), fatigue, bone pain, headaches, insomnia; on tetany Chvostek’s & Trousseau’s signs; on severe hypocalcemia arrhythmias, and spasm of the upper part of the airways (laryngospasm) or the bronchi may occur].
· Causes: thyroidectomy (surgical removal of parathyroids) or trauma of the parathyroids during surgery or compromise of their blood flow during surgery, or trauma of the parathyroids; autoimmune invasion & destruction of the parathyroids (the most common non-surgical cause; may be part of autoimmune polyendocrine syndromes); DiGeorge syndrome (absence or dysfunction of parathyroid glands); magnesium deficiency; defect in the calcium receptor; idiopathic; familial e.g. Barakat syndrome (hypoparathyroidism, sensorineural deafness, renal disease) or autoimmune polyglandular failure syndrome type 1 (APS – I; familial hypoparathyroidism & other endocrine diseases e.g. adrenal insufficiency).
· Biochemistry in hypoparathyroidism: low PTH (parathormone; parathyroid hormone), low calcitriol, low calcium, high phosphates.
· Pseudohypoparathyroidism: it is characterized by end-organ resistance to the effects of PTH. PTH binds to the PTH receptor, which, in turn, activates cAMP through guanine nucleotide regulatory proteins (Gs). These proteins consist of alpha, beta, and gamma subunits. Pseudohypoparathyroidism is classified into types I and II. Type I is further subdivided into Ia, Ib, and Ic.
· a) Type Ia pseudohypoparathyroidism. It results from a decrease in the Gs-alpha protein. This disorder comprises the biochemical features of pseudohypoparathyroidism along with the following somatic features of Albright hereditary osteodystrophy (AHO): short stature, mental retardation, obesity, round-shaped face, brachymetacarpia, brachymetatarsia, and subcutaneous bone formation.
· Laboratory findings in AHO include hypocalcemia, hyperphosphatemia (with normal or high PTH levels), and low calcitriol (1,25(OH)2D3 - the active form of vitamin D made in the kidney). Vitamin D may be decreased because of inhibition by elevated levels of phosphorus and by decreased PTH stimulation of 25-hydroxyvitamin D 1-alpha-hydroxylase. The low calcitriol levels, in turn, may cause resistance to the hypercalcemic effects of PTH in the bone. The gene for the Gs-alpha protein is located on chromosome 20. Some family members carry the mutation and display the AHO phenotype but do not have pseudohypoparathyroidism. This is termed pseudo-pseudohypoparathyroidism.
· b) Type Ib pseudohypoparathyroidism. The patients do not present with the somatic features of AHO. These patients have normal Gs-alpha protein, with hormonal resistance to PTH—an impaired cAMP response to PTH, suggesting that the defect lies on the receptor.
· c) Type Ic pseudohypoparathyroidism. Patients present with resistance to multiple hormonal receptors. However, Gs-alpha protein expression is normal.
· d) Type II pseudohypoparathyroidism. PTH raises cAMP normally but fails to increase levels of serum calcium or urinary phosphate excretion, suggesting that the defect is located downstream of the generation of cAMP. These patients present with hypocalcemia, hypophosphaturia, and elevated immunoreactive PTH (iPTH) levels. These findings also occur in vitamin D deficiency, but in patients with a vitamin D deficiency, all parameters return to normal after vitamin D administration.
· Pseudohypoparathyroidism – characteristics & laboratory investigation:
· a) Type 1a: characteristic phenotypic appearance (Albright’s hereditary osteodystrophy; short 4th & 5th metacarpals, rounded facies), usually autosomal dominant; PTH resistance. Appearance: skeletal defects. Biochemistry: high PTH, low calcitriol, low calcium, high phosphates. Gene defect from mother (GNAS1).
· b) Type 1b: lacks the physical appearance of type 1 a but is biochemical similar. Biochemistry: high PTH, low calcitriol, low calcium, high phosphates. Gene defect from mother (GNAS1 & STX16).
· c) Type 2: lacks the physical appearance of type 1a; normal cAMP response to PTH stimulation, despite the inherent abnormality in calcium regulation. Biochemistry: high PTH, low calcitriol, low calcium, high phosphates. Note: calcitriol or 1,25-dihydroxycholecalciferol or 1,25-dihydroxy vitamin D3, is the hormonally active metabolite of vitamin D.
· Pseudopseudohypoparathyroidism (pseudoPHP): an inherited disorder; the individual has the phenotypic appearance of pseudohypoparathyroidism type 1a but is biochemically normal. It is sometimes considered a variant of Albright’s hereditary osteodystrophy. The GNAS1 gene involved in both pseudohypoparathyroidism type 1a and pseudopseudohypoparathyroidism is significantly affected by imprinting. Appearance: skeletal defects.
· Biochemistry: normal PTH, calcitriol, calcium, and phosphates.
· Hyperparathyroidism: is overactivity of the parathyroid glands resulting in excess production of parathyroid hormone (PTH). This hormone regulates calcium and phosphate levels and helps to maintain these levels. Classification:
· a) Primary hyperparathyroidism results from a hyperfunction of the parathyroid glands themselves. There is over-secretion of PTH due to a parathyroid adenoma, parathyroid hyperplasia, or rarely, a parathyroid carcinoma. Clinically, this disease because the symptoms of hypercalcemia are often characterized by the quartet ‘stones, bones, groans, thrones, and psychic overtones’ referring to the presence of kidney stones, hypercalcemia, constipation and peptic ulcer disease, and depression, respectively. In a minority of cases, this occurs as part of multiple endocrine neoplasia (MEN) syndrome, either type 1 (mutation in the gene MEN1) or type 2a (mutation in the gene RET). Also, patients with bipolar disorder who are receiving long-term lithium treatment are at increased risk for hyperparathyroidism (however, only a few patients have significantly elevated levels of parathyroid hormone and clinical symptoms of hyperparathyroidism). Lithium-associated hyperparathyroidism is usually caused by a single parathyroid adenoma.
· b) Secondary hyperparathyroidism is due to physiological (i.e., appropriate) secretion of PTH by the parathyroid glands in response to hypocalcemia (low blood calcium levels). The most common causes are vitamin D deficiency (caused by lack of sunlight, or diet deficient in vitamin D or by malabsorption, e.g., intestinal problems) and chronic renal (kidney) failure. Lack of vitamin D leads to reduced calcium absorption by the intestine leading to hypocalcemia and increased parathyroid hormone secretion. This increases bone resorption. In chronic kidney disease (CKD) the problem is more specifically failure to convert vitamin D to its active form in the kidney (calcitriol (1,25(OH)2D3). The bone disease in secondary hyperparathyroidism caused by renal failure is called renal osteodystrophy.
· c) Tertiary hyperparathyroidism is seen in patients with long-term secondary hyperparathyroidism which eventually leads to hyperplasia of the parathyroid glands and a loss of response to serum calcium levels. It most often is seen in patients with chronic renal failure and is an autonomous activity.
· d) Quaternary and quinary hyperparathyroidism: these are rare conditions that may be observed after surgical removal of primary hyperparathyroidism when it has led to kidney damage that now again causes a form of secondary (quaternary) hyperparathyroidism that may itself result in autonomy (quinary) hyperparathyroidism. Also, quaternary hyperparathyroidism may ensue from hungry bone syndrome after parathyroidectomy (*).
· Signs & symptoms: these depend entirely on whether the hyperparathyroidism is primary or secondary:
· i) In primary hyperparathyroidism about 50% of patients have no symptoms, and the problem is picked up as an incidental finding via a raised calcium or characteristic X-ray appearance. Many other patients only have non-specific symptoms. Symptoms directly due to hypercalcemia are relatively rare, being more common in patients with malignant hypercalcemia. If present, common manifestations of hypercalcemia include weakness and fatigue, depression, bone pain, muscle soreness (myalgias), decreased appetite, nausea & vomiting, constipation, polyuria, polydipsia, cognitive impairment, kidney stones, and osteoporosis. Bone resorption may cause signs such as racquet nails (trachyonychia; the nail plate is flattened, the end of the thumb is widened and flattened, and the distal phalanx is abnormally short). Parathyroid adenomas are very rarely detectable on clinical examination. Surgical removal of a parathyroid tumor will eliminate the symptoms in most patients.
· ii) In secondary hyperparathyroidism the parathyroid gland is behaving normally; clinical problems are due to bone resorption and manifest as bone syndromes such as rickets (in children), osteomalacia, and renal osteodystrophy. Osteomalacia is the softening of the bones caused by defective bone mineralization secondary to inadequate levels of available phosphate & calcium, or because of overactive resorption of calcium from the bone which can be caused by hyperparathyroidism that causes hypercalcemia. Renal osteodystrophy or chronic kidney disease-mineral and bone disorder (CKD – MBD) is a bone problem characterized by bone mineralization deficiency, that is a direct result of the electrolyte & endocrine derangements that accompany chronic kidney disease. It is thought to be the result of hyperparathyroidism, secondary to hyperphosphatemia combined with hypocalcemia (exacerbated by the low levels of activated vitamin D3 as a result of the damaged kidneys' inability to convert vitamin D3 into its active form, calcitriol), both of which are due to decreased excretion of phosphate by the damaged kidney.
· Imaging tests: a technetium sestamibi scan is a procedure in nuclear medicine which is performed to identify hyperparathyroidism (or parathyroid adenoma). It may also locate an ectopic parathyroid adenoma.
· (*) Parathyroidectomy is the treatment of choice in patients with primary hyperparathyroidism (PHPT). This disease affects calcium metabolism at the level of bone tissue and renal tubules, resulting in hypercalcemia, often asymptomatic, associated with hypophosphatemia and hypomagnesemia. Sudden suppression of parathyroid hormone (PTH), caused by successful parathyroidectomy, in patients with high preoperative levels of PTH and hypercalcemia from enhanced bone turnover, may induce severe postoperative hypocalcemia that may lead to symptoms of tetany. This relatively uncommon condition is known as ‘hungry bone syndrome’ (HBS) because it is believed to be due mainly to enhanced bone formation.
· Racquet nails (image):
· Hyperparathyroidism – Laboratory investigation: the gold standard of diagnosis is the parathyroid immunoassay. Once an elevated PTH has been confirmed, the goal of diagnosis is to determine whether the hyperparathyroidism is primary or secondary in origin by obtaining a serum calcium levels (or the ionized calcium levels that are not affected by low albumin levels).
· a) In primary hyperparathyroidism parathyroid hormone (PTH) is high; serum calcium is high; alkaline phosphatase (ALP) is elevated or inappropriately normal; serum phosphate is abnormally low in about 50% of cases.
· b) In secondary hyperparathyroidism parathyroid hormone (PTH) is high; serum calcium is low or normal; serum phosphate is elevated in renal disease. The combination of normal serum calcium, low phosphate, and elevated alkaline phosphatase is suggestive of disturbed vitamin D metabolism.
· If creatinine and BUN are also increased, the problem probably lies with the renal production of 1,25-OHD.
· If liver function tests are abnormal or serum albumin is low, the problem may be a low 25-OHD level due to liver disease or malnutrition.
· When 1,25-OHD levels are low, either due to deficient vitamin D stores or renal disease, the parathyroid glands release more PTH to try to synthesize more 1,25-OHD. Since this cannot happen, increased PTH promotes calcium absorption from skeletal bone, which may result in severe bone disease. Children develop rickets, while adults develop osteomalacia.
· PTH assay: Typically PTH levels vary significantly over time in the affected patient and must be retested several times to see the pattern. The currently accepted test for PTH is ‘Intact PTH’ which is intended to detect only relatively intact and biologically active PTH molecules. Older tests often detected other, inactive fragments. However, even ‘Intact PTH’ may be inaccurate in patients with renal dysfunction.
· In cases of primary hyperparathyroidism or tertiary hyperparathyroidism elevated PTH leads to increased serum calcium due to: increased bone resorption, allowing the flow of calcium from bone to blood; reduced kidney clearance of calcium; and increased intestinal calcium absorption.
· Parathyroid hormone-related protein (PTHrP): PTH-related peptide is produced by some cancers, including those of the lung, breast, head, neck, bladder, gastrointestinal tract, and ovaries, as well as leukemia and lymphoma. High levels of PTH-related protein may be the cause of elevated calcium levels in about 2/3 of cancer patients (humoral hypercalcemia of malignancy (HHM)). Since PTHrP has the same N-terminal as PTH (parathyroid hormone), it can bind to the same receptor and stimulate the action of PTH. This results in bone resorption and calcium resorption in the kidneys but has minimal effect on calcium absorption in the intestines. No detectable (or minimal) PTH-like protein is normal. Women who are breastfeeding may have detectable PTH-related protein values. Increased levels of PTH-related protein with high blood calcium level is usually caused by cancer.
Vitamin D
· Vitamin D is the hormone that enhances intestinal absorption of calcium and ensures healthy bone formation. The best way to obtain vitamin D is through direct exposure of the skin to sunlight because ultraviolet B rays stimulate the skin to synthesize vitamin D3. Some people do not have adequate exposure to sunlight and are at higher risk of developing vitamin D deficiency. Examples include individuals who: live in latitudes >35o from the equator; live in heavily polluted cities; belong to cultures that require clothing that covers their entire body; are institutionalized or homebound; have dark skin; apply sunscreen compulsively; are elderly (decreased skin synthesis), and are obese (vitamin accumulates in fat). Individuals ingesting a balanced diet obtain 200 to 300 IU of vitamin D per day. In 2010, The Institute of Medicine recommended a vitamin D daily intake of 600 IU for children and adults < 70 years and 800 IU for adults older than 70 years. The Endocrine Society recommends a higher vitamin D daily intake of 1,500 to 2,000 IU daily. In the United States, most vitamin D supplements contain vitamin D3 while prescriptions consist of vitamin D2.
· Calcitriol or 1,25-dihydroxycholecalciferol or 1,25-dihydroxy vitamin D3 is the hormonally active metabolite of vitamin D.
· Cholecalciferol (colecarciferol, vitamin D3) is one of the five forms of vitamin D.
· 7 – Dehydrocholesterol (provitamin D3) is the precursor of cholecalciferol. Within the epidermal layer of skin, it undergoes an electrocyclic reaction as a result of UVB radiation, resulting in the opening of the vitamin precursor B-ring through a conrotatory pathway. Following this, it finally isomerizes to form cholecalciferol (vitamin D3). Cholecalciferol is then hydroxylated in the liver to become calcifediol (25-hydroxyvitamin D3; also known as calcidiol, 25-hydroxycholecalciferol; abbreviated 25(OH)D). Calcifediol is then hydroxylated in the kidney, under the regulation of parathyroid hormone (PTH), and becomes calcitriol (1,25-dihydroxy vitamin D3) or active vitamin D3.
· Therefore, normal vitamin D metabolism is dependent on sunlight exposure, intestinal absorption, and liver and kidney function. Vitamin D malabsorption may be associated with several GI disorders including Crohn's disease, celiac disease, and pancreatic insufficiency. As vitamin D levels fall, the initial compensatory mechanism is increased PTH secretion, which stimulates the kidneys to increase phosphate excretion and decrease calcium excretion. Blood calcium levels remain normal until the very late stages of vitamin D deficiency.
· Alkaline phosphatase is usually elevated in response to the effect of PTH on calcium absorption from the bone. The combination of normal serum calcium, low phosphate, and elevated alkaline phosphatase is suggestive of disturbed vitamin D metabolism.
· If creatinine and BUN are also increased, the problem probably lies with the renal production of 1,25-OHD.
· If liver function tests (LFTs) are abnormal or serum albumin is low, the problem may be a low 25-OHD level due to liver disease or malnutrition.
· When 1,25-OHD levels are low, either due to deficient vitamin D stores or renal disease, the parathyroid glands release more PTH to try to synthesize more 1,25-OHD. Since this cannot happen, increased PTH promotes calcium absorption from skeletal bone, which may result in severe bone disease. Children develop rickets, while adults develop osteomalacia. Osteomalacia may present as a diffuse, dull, aching pain affecting many areas of the body including ribs and sternum.
· Vitamin D deficiency is a significant risk factor for osteoporosis, bone loss, weakness, and fracture in the elderly.
· Some patients taking long-term antiepileptic drug therapy to develop a syndrome of low plasma 25-OHD, intestinal malabsorption of calcium, a slight decrease in plasma calcium, secondary hyperparathyroidism, and cortical osteopenia.
· In one study of inpatients on a medical ward, the most common disorders associated with vitamin D deficiency were anticonvulsant therapy, renal dialysis, nephrotic syndrome, and winter season, and also contributory factors were liver cirrhosis, malabsorption, and glucocorticoid therapy.
· Both 25-OHD and 1,25-OHD can be measured to assess vitamin D status. There are two principal forms of vitamin D: D2 and D3. Many of the currently available assays measure and report on both vitamin D2 and D3 metabolites. This can be useful in studies evaluating the contribution of vitamin D2 and D3 to overall vitamin D status. 25-hydroxyvitamin D (25(OH)D) is the primary circulating form of vitamin D. Thus, the total serum 25(OH)D level is currently considered the best indicator of vitamin D supply to the body from cutaneous synthesis and nutritional intake. One exception is that 25(OH)D levels do not indicate clinical vitamin D status in patients with chronic renal failure or type 1 vitamin D-dependent rickets or when calcitriol (1,25-dihydroxy vitamin D) is used as a supplement. 25-OHD is the preferred test for patients with normal renal function. Measurement of 1,25-OHD should be restricted to patients with renal disease.
· Interpretation of 25(OH)D can be challenging owing to wide variability in patient’s weight, ethnicity, assays, laboratory procedures, and validation of reference ranges.
· The reference range for 25-OHD (25-hydroxyvitamin D3 or 25-hydroxycholecalciferol; also known as calcidiol or calcifediol; abbreviated 25(OH)D) is:
· a) Deficiency: vitamin 25-OHD < 12 ng/ml.
· b) Insufficiency: vitamin 25-OHD 12 – 30 ng/ml.
· c) Adequate levels: vitamin 25-OHD 30 – 79 ng/ml.
· d) Toxicity possible: vitamin 25-OHD > 80 ng/ml.
· According to the Mayo Clinic the reference range for 25-OHD is:
· a) Severe deficiency (could be associated with rickets in children & osteomalacia in adults): vitamin 25-OHD < 1o ng/ml.
· b) Mild to moderate deficiency (may be associated with increased risk for osteoporosis or secondary hyperparathyroidism): vitamin 25-OHD 10 – 25 ng/ml.
· c) Optimum levels (in the normal population): vitamin 25-OHD 25 – 79 ng/ml.
· d) Toxicity possible: vitamin 25-OHD > 80 ng/ml (80 ng/ml is the lowest reported level associated with toxicity in patients without primary hyperparathyroidism, and with normal renal function).
· According to Medscape, the reference range for 25-OHD is:
· a) Deficiency: vitamin 25-OHD < 20 ng/ml (50 nmol/L).
· b) Insufficiency: vitamin 25-OHD 21 – 29 ng/ml (52-72 nmol/L).
· c) Sufficiency: vitamin 25-OHD 30 – 149 ng/ml (75 – 374 nmol/L).
· d) Toxicity: vitamin 25-OHD > 150 ng/mL (374 nmol/L).
· Individuals with intense sun exposure have a mean 25- hydroxyvitamin D level of 36 ng/mL and a range of 20 – 70 ng/mL.
· In 2010, the Institute of Medicine endorsed a 25-OHD concentration of 20 ng/mL as being adequate to prevent rickets and osteomalacia.
· In the United States, 30 to 35% of individuals have vitamin D levels below 20 ng/mL. However, 21% of individuals with 25-OHD levels between 20 – 30 ng/mL have increased osteoid volume. Most experts believe that the optimal concentration of 25-OHD is at least 30 ng/mL because this is the threshold for the elevation of parathyroid hormone. The American Association of Clinical Endocrinology recommends that physicians target a 25-OHD concentration between 30 and 50 ng/mL.
Thyroid disorders
· Thyroid function tests (TFTs): is a collective term for blood tests used to check the function of the thyroid. TFTs may be requested if a patient is thought to suffer from hyperthyroidism (overactive thyroid) or hypothyroidism (underactive thyroid) or to monitor the effectiveness of either thyroid suppression or hormone replacement therapy. It is also requested routinely in conditions linked to thyroid diseases, such as atrial fibrillation (AF; a heart arrhythmia) and anxiety disorders. A TFT panel typically includes thyroid hormones such as thyroid stimulating hormone (TSH; also called thyrotropin) and thyroxine (T4), and triiodothyronine (T3) depending on local laboratory policy. Thyroxine (T4) is released from the thyroid gland along with small amounts of triiodothyronine (T3) and thyroglobulin under the guidance of thyroid-stimulating hormone (TSH). Secretion of TSH is principally regulated by circulating levels of thyroid hormones (via a negative feedback loop) and hypothalamic thyrotropin-releasing hormone (TRH). More than 99% of the thyroid hormones are bound to proteins, including thyroid-binding globulin (TBG), thyroxine-binding pre-albumin, and albumin. Less than 1% is free, unbound hormones that make up the biologically active fraction, and they are not generally influenced by thyroid-binding protein abnormalities. Thyroxine is tightly bound to TBG, whereas T3 is less tightly bound to TBG but more tightly bound to thyroxine-binding pre-albumin and albumin. The majority of serum T3 levels (over 75%) come from the peripheral conversion of T4. In nonthyroidal illness, T4 conversion to T3 is reduced and conversion to reverse T3 (rT3) is enhanced.
· Ancillary tests, such as those for thyroid-stimulating immunoglobulin (TSI) levels and antithyroid antibody levels are useful in the management of Graves' disease and Hashimoto's disease, respectively. The most common antithyroid antibodies (antimicrosomal/ peroxidase and antithyroglobulin) are highly organ-specific and organ-sensitive. The antimicrosomal antibodies, directed primarily against membrane-bound thyroid peroxidase, are most useful in diagnosing Hashimoto's thyroiditis. Tests for these antibodies may also be positive in Graves' disease.
· Hyperthyroidism: a sensitive serum thyroid-stimulating hormone (TSH) assay should be used. Low serum TSH, especially if below the reference range, but above 0.10 mU/L, is not specific for hyperthyroidism, as it may also occur in non – thyroidal illness, or with the use of some drugs. A subnormal TSH should trigger the measurement of free thyroxine (FT4). If it is not elevated, then free triiodothyronine (FT3) should also be measured to identify T3 thyrotoxicosis.
· Thyroid autoantibodies [thyroid peroxidase antibodies (TPOAb), TSH – receptor antibodies (TRAb), or thyroid-stimulating immunoglobulins (TSI)] may be measured in selected cases.
· Note: antithyroid antibodies are negative in about 10% of cases of Grave’s disease.
· Radioactive isotope uptake – thyroid scanning with radiolabeled iodine (I123) may give information (e.g., diffuse uptake in Graves vs. one or more ‘hot’ nodules in toxic nodular hyperthyroidism).
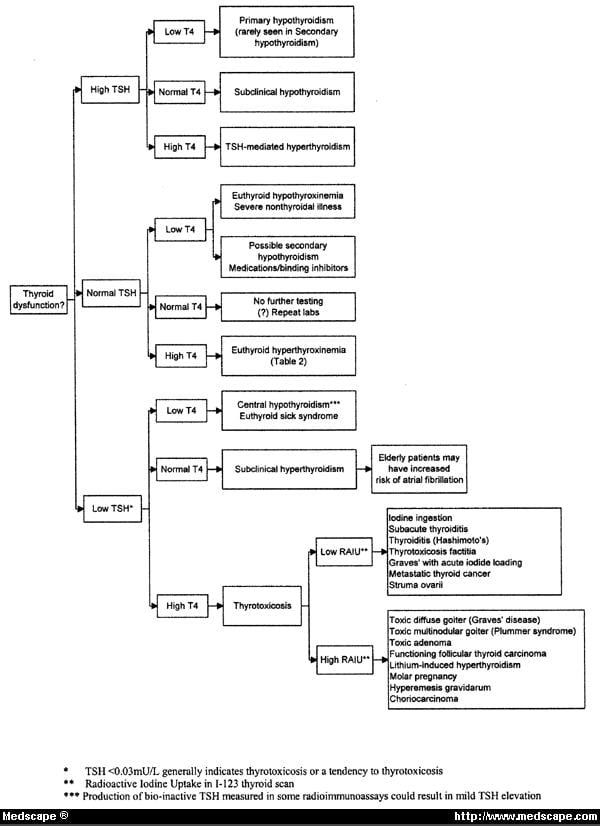
· Interpretation of TFTs (thyroid function tests) on hyperthyroidism:
· a) Low TSH; raised FT3 or FT4 – common causes: primary hyperthyroidism – Grave’s disease, multinodular goiter, toxic nodule. Relative common causes with low radio-iodine uptake: transient thyroiditis (postpartum, post–viral, thyroiditis De Quervain’s).
· Rare causes with low radioiodine uptake: thyroxine (T4) ingestion, ectopic thyroid tissue, iodine-induced, amiodarone drug therapy.
· Rare causes with positive pregnancy test: gestational thyrotoxicosis with hyperemesis gravidarum; hydatiform mole. Rare causes: familial TSH receptor mutation.
· b) Low TSH; normal FT3 or FT4 – common causes: subclinical hyperthyroidism; thyroxine (T4) ingestion. Rare causes: steroid therapy; dopamine and dobutamine infusion; non – thyroidal illness.
· c) Low or normal TSH; low FT3 or FT4 – common causes: non – thyroidal illness; recent treatment for hyperthyroidism. Rare causes: pituitary disease; congenital TSH or TRH deficiencies.
· Hypothyroidism: to diagnose primary hypothyroidism we need to measure TSH and FT4. When TSH is > 10 mU/L and FT4 below the reference range, then the diagnosis is overt hypothyroidism, and the patient needs treatment with thyroid replacement therapy. On secondary hypothyroidism we have low, within or mildly elevated TSH combined with a low FT4; FT4 and sometimes anterior pituitary hormone tests are necessary to differentiate this from non – thyroidal illness.
· Interpretation of TFTs (thyroid function tests) on hypothyroidism:
· a) Raised TSH; low FT4 or FT3
· Common causes: chronic autoimmune thyroiditis; after radio-iodine; after thyroidectomy; transient thyroiditis – hypothyroid phase.
· Rare causes – anti-TPO negative: after external beam radiotherapy to the neck; drugs (amiodarone, lithium, interferons, interleukin – 2); iodine deficiency; amyloid goiter.
· Congenital causes: thyroid dysgenesis; iodine transport defects; TSH – receptor defects; TSH – resistance.
· b) Raised TSH; normal FT4 or FT3
· Common causes: subclinical autoimmune hypothyroidism.
· Rare causes: drugs (amiodarone, sertraline (an SSRI antidepressant), cholestyramine); recovery phase after a non – thyroidal illness; heterophile (interfering antibody).
· Congenital causes: TSH – receptor defects; TSH – resistance.
· c) Normal or raised TSH; raised FT4 or FT3
· Rare causes: amiodarone; interfering antibodies; familial; TSH – secreting pituitary tumor; acute psychiatric illness.
· Subclinical thyroid disease: it is common; in the USA subclinical hypothyroidism occurs in 4 – 8.5% of the general population, and up to 20% of women > 60 years old; about 2% of the general population has subclinical hyperthyroidism (low serum TSH; normal FT4 and FT3 in the absence of non – thyroidal illness or relevant drug therapy) that may increase the risk for atrial fibrillation (AF; an abnormal heart rhythm) and CVD (cardiovascular disease)).
· Algorithm for interpretation of thyroid function tests (TFTs):
{kind=link}
{kind=link}
{kind=link}
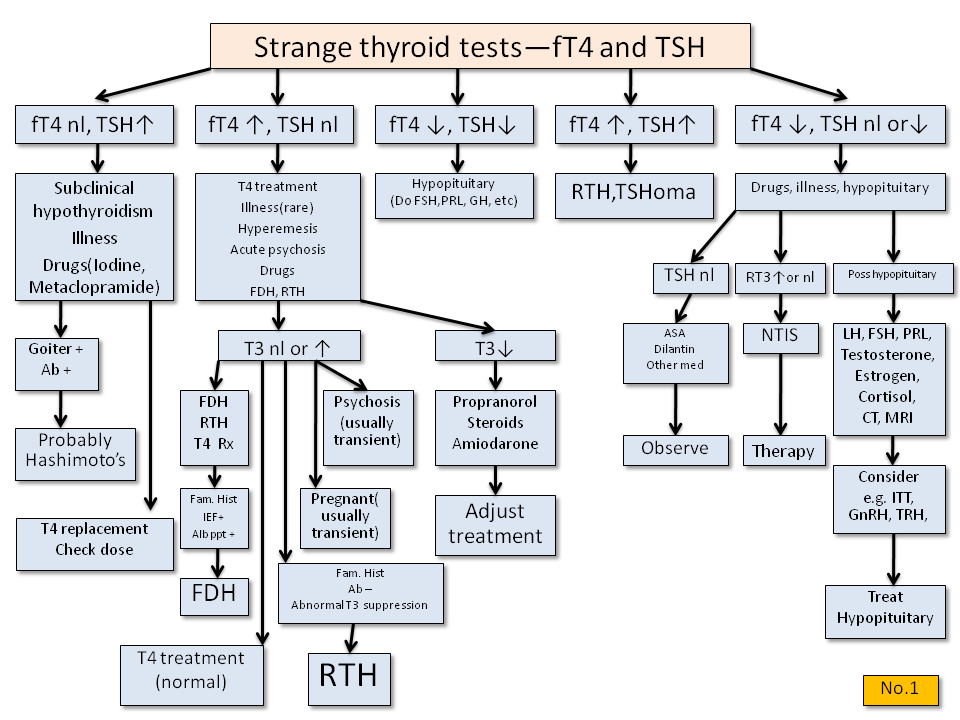
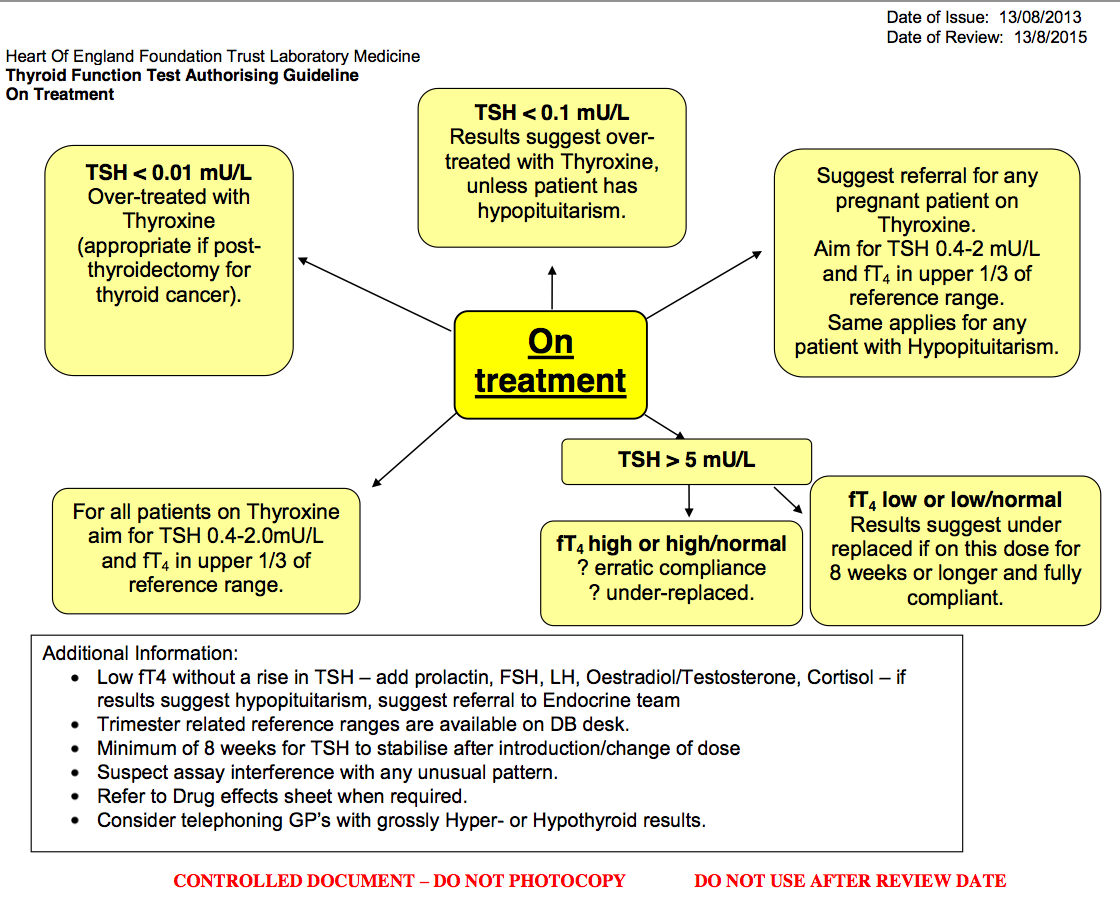
· Interpretation of thyroid function tests on patients taking thyroxine (T4):
{kind=link}
· Calcitonin (also known as thyrocalcitonin): a 32-amino acid linear polypeptide hormone that is produced primarily by the parafollicular cells (C-cells) of the thyroid. It acts to reduce blood calcium (Ca2+), opposing the effects of parathyroid hormone (PTH). Both increased calcitonin secretion and increased calcitonin activity are relatively short-lived, lasting only a few days. As a result, patients with chronically high serum calcium concentrations (hypercalcemia) do not have elevated serum calcitonin concentrations.
· Calcitonin may be used diagnostically as a tumor marker for medullary thyroid cancer (a cancer of the parafollicular cells that secretes large quantities of calcitonin), in which high calcitonin levels may be present and elevated levels after surgery may indicate recurrence.
· Medullary thyroid cancer may be part of the MEN (multiple endocrine neoplasias) 2 syndrome. Patients with medullary thyroid carcinoma have high serum calcitonin concentrations, but normal serum calcium concentrations. It may even be used on biopsy samples from suspicious lesions (e.g., swollen lymph nodes) to establish whether they are metastasis of primary cancer.
· Increased levels of calcitonin have also been reported for various other conditions, including C-cell hyperplasia; non-thyroidal oat cell carcinoma, non-thyroidal small cell carcinoma and other non-thyroidal malignancies; lung cancer; VIPoma; insulinoma; acute and chronic renal failure; hypercalcemia; hypergastrinemia and other gastrointestinal disorders; and pulmonary disease.
· Note: synthetic analogs of calcitonin (such as salmon’s intranasal calcitonin) are of particular interest in the treatment of osteoporosis. It may also be used to treat hypercalcemia (increased blood calcium) and Paget's disease of the bone.
Adrenal disorders
· ACTH stimulation test (Synacthen test; tetracosactide test; Cosyntropin test): a test that assesses the functioning of adrenal gland stress response by measuring the adrenal response to ACTH (adrenocorticotropic hormone; a hormone produced by the anterior pituitary gland that stimulates the adrenal glands to release cortisol and DHEAS (dehydroepiandrosterone)). In the ACTH stimulation test, a small amount of synthetic ACTH is injected, and the amount of cortisol and sometimes of aldosterone is measured. It is used to diagnose primary and secondary adrenal insufficiency and Addison’s disease. It distinguishes whether the cause is adrenal (low cortisol and aldosterone production), or pituitary (low ACTH production).
· Versions οf the test:
· a) Low dose short test: 1 μg/kg of ACTH is injected.
· b) Conventional – short dose test: 250 μg of ACTH is injected. The previous 2 tests last about 1 hour. c) Prolonged – stimulation test (long conventional dose test). It can last up to 48 hours and can differentiate primary, secondary and tertiary adrenal insufficiency.
· Preparation: the person must fast at least 6 hours before the test, and should not take glucocorticoids, adrenal extract supplements, spironolactone (a diuretic), contraceptives, licorice (a herb), estrogen, androgens (including DHEA), and progesterone. Moreover, the person should not be under stress or have been administered radioisotope scans recently. Women should be in the 1st week of their menstrual cycle. Also, for aldosterone measurement, salt consumption should be reduced.
· Procedure: cortisol and ACTH are measured at baseline (time = 0). Next, synthetic ACTH is injected IM or IV. About 20 ml of heparinized venous blood is collected at 30 and 60 min after ACTH injection to measure cortisol. ACTH is kept in ice and sent immediately to the lab.
· Interpretation:
· I) ACTH stimulation test.
· a) In healthy people the cortisol level doubles from a baseline of 20 – 30 μg/dl within 60 min (e.g., if serum cortisol was 25 μg/dl before the stimulation, it should reach at least 50 μg/dl).
· b) Primary adrenal insufficiency & Addison’s disease: baseline cortisol is well below 10 μg/dl and rises no more than 25%. Cortisol values less than doubles in stimulation.
· c) Secondary adrenal insufficiency: ACTH may dramatically stimulate cortisol from the low baseline value, and stimulation may result even in a greater than 14 fold increase in a serum concentration over 30 min. However, more typical serum cortisol levels will double or triple from baseline. If secondary adrenal insufficiency is diagnosed, the insulin tolerance test (ITT) or the corticotropin-releasing hormone (CRH) stimulation test can be used to distinguish between the hypothalamic (tertiary) and pituitary (secondary) cause.
· II) ACTH plasma test. It should be given simultaneously with the ACTH stimulation.
· a) A normal ACTH value is in the upper third of the range (range 10 – 60 ng/L).
· b) Primary adrenal insufficiency & Addison’s disease: ACTH is high, either at the top or above the range. In Addison’s disease, ACTH may reach the hundreds.
· c) Secondary adrenal insufficiency: ACTH is low, usually below 35, but in most people with secondary adrenal insufficiency it will fall within the range limit. In some cases, low ACTH may be caused by low CRH in the hypothalamus. In head injury, we may have separate ACTH and CRH impairment.
· III) Aldosterone stimulation. The ACTH test is used occasionally to help to determine if the primary (hyperreninemic) or secondary (hyporeninemic) hypoaldosteronism is present. The amount of synthetic ACTH in the stimulation test has a stimulatory effect in aldosterone production.
· a) On normal individuals, aldosterone should double from a correct base value (around 20 ng/dl). The person must fast from salt 24 hours before the test and should sit upright for a blood draw.
· b) Primary aldosterone deficiency: the aldosterone response (doubling) is blunted or absent. The base value rises less than double from the base value. This indicates primary hyperreninemic hypoaldosteronism with low sodium, high potassium, and high rennin. This is indicatory of primary adrenal insufficiency or Addison’s disease.
· c) Secondary aldosterone deficiency: aldosterone response of several factors from a low base value and usually doubling or quadrupling from a low base aldosterone value is what is seen. This indicates secondary hyporeninemic hypoaldosteronism with low sodium, low potassium, and low rennin. A result of doubling or more of aldosterone may help in tandem with an ACTH stimulation test that doubled or more confirms a diagnosis of secondary adrenal insufficiency. In rare cases, an aldosterone stimulation where aldosterone has not doubled, with the presence of low potassium, low renin, and low ACTH, indicates insufficiency of aldosterone production from prolonged lack of rennin.
· IV) Other hormones that rise in the ACTH stimulation test: progesterone, LH (luteinizing hormone), 21 – hydroxylase, DHEA (dehydroepiandrosterone), and DHEA – S (dehydroepiandrosterone sulfate).
· Diagnostic chart for ACTH stimulation test:
· a) Primary adrenal insufficiency, including Addison’s disease: high CRH; high ACTH; high DHEA; high DHEA – S; low cortisol (values less than doubles in ACTH stimulation test); low aldosterone; high renin (hyperreninemic hypoaldosteronism); low Na+ (sodium), high K+ (potassium).
· Primary adrenal insufficiency – common causes: a tumor of the adrenal (adenoma), stress, antibodies, environment, Addison’s disease, injury, surgical removal.
· b) Secondary (pituitary) adrenal insufficiency: high CRH (only if CRH production in the hypothalamus is intact); low ACTH; low DHEA; low DHEA – S; low cortisol (values doubles or more in ACTH stimulation test); low aldosterone; low renin (hyporeninemic hypoaldosteronism); low Na+ (sodium), low K+ (potassium).
· Secondary (pituitary) adrenal insufficiency – common causes: a tumor of the pituitary (adenoma), antibodies, environment, head injury, surgical removal (usually because of macroadenoma), Sheehan’s syndrome (hypopituitarism caused by ischemic necrosis due to blood loss and hypovolemic shock during and after childbirth).
· c) Tertiary (hypothalamus) adrenal insufficiency (automatically includes diagnosis of secondary (hypopituitarism) adrenal insufficiency): low CRH; low ACTH; low DHEA; low DHEA – S; low cortisol (values doubles or more in ACTH stimulation test); low aldosterone; low renin (hyporeninemic hypoaldosteronism); low Na+ (sodium), low K+ (potassium).
· Tertiary (hypothalamus) adrenal insufficiency – common causes: a tumor of the hypothalamus (adenoma), antibodies, environment, head injury.
· Diagnostic algorithms for adrenal insufficiency:
{kind=link}
{kind=link}
· Addison’s disease: a rare, chronic endocrine system disorder in which the adrenal glands do not produce sufficient steroid hormones (glucocorticoids and often mineralocorticoids).
· It is characterized by some relatively nonspecific symptoms such as abdominal pain and weakness.
· On physical examination, the following clinical signs may be noticed: hypotension, +_ orthostatic hypotension; and hyperpigmentation (darkening of the skin, including areas not exposed to the sun, such as skin creases (e.g. of the palms), nipple, and the inside of the cheek (buccal mucosa); also, old scars may darken.
· Routine laboratory investigations may show the following: hypercalcemia, hypoglycemia, hyponatremia, hyperkalemia, eosinophilia and lymphocytosis, and metabolic acidosis.
· Under certain circumstances of stress (such as surgery), symptoms may progress to an Addisonian crisis, a severe illness which may include severe hypotension and coma.
· Causes:
· Primary adrenal insufficiency. Autoimmune adrenalitis with autoimmune destruction of the adrenal cortex (immune reaction against the enzyme 21 – hydroxylase) is the most common cause of Addison's disease in the industrialized world. This may be isolated or in the context of the autoimmune polyendocrine syndrome (APS type 1 or 2), in which other hormone-producing organs, such as the thyroid and pancreas may also be affected.
· Adrenal destruction is also a feature of adrenoleukodystrophy (ALD; a disorder of peroxisomal fatty acid beta-oxidation which results in the accumulation of very-long-chain fatty acids in tissues throughout the body, especially affected areas are the myelin in the CNS, the adrenal cortex, and the Leydig cells in the testes), and when the adrenal glands are involved in metastasis, hemorrhage (e.g. in Waterhouse – Friderichsen syndrome (most commonly caused by the meningococcus Neisseria meningitides) or antiphospholipid syndrome) and particular infections (tuberculosis, histoplasmosis, coccidiomycosis), or the deposition of abnormal protein in amyloidosis. It may also be caused by certain diseases or various rarer causes. The use of high-dose steroids for more than a week begins to produce suppression of the patient's adrenal glands because the exogenous glucocorticoids suppress hypothalamic corticotropin-releasing hormone (CRH) and pituitary adrenocorticotropic hormone (ACTH). With prolonged suppression, the adrenal glands atrophy and can take months to recover full function after discontinuation of the exogenous glucocorticoid. During this recovery time, the patient is vulnerable to adrenal insufficiency during times of stress, such as illness. Abrupt corticosteroid withdrawal may cause Addison’s.
· Other rare causes include adrenal dysgenesis (genetic), interruptions in the delivery of cholesterol (Smith – Lemli – Opitz syndrome and abetalipoproteinemia), congenital adrenal hyperplasia (21 – hydroxylase, 17 alpha-hydroxylase, 11 beta-hydroxylase and 3 beta-hydroxysteroid dehydrogenase), lipoid CAH (congenital adrenal hyperplasia (deficiency of StAR and mitochondrial DNA mutations). Finally, some medications interfere with steroid synthesis enzymes (e.g., ketoconazole), while others accelerate the typical breakdown of hormones by the liver (e.g., rifampicin and phenytoin).
· Hyperaldosteronism: the excessive levels of aldosterone that may be independent of the renin-angiotensin axis (primary hyperaldosteronism) or may be due to high renin levels (secondary hyperaldosteronism).
· In hypertensive patients, the prevalence is 5 – 10% (in patients with resistant hypertension hyperaldosteronism should be investigated).
· Mechanism: excessive aldosterone acts at the distal renal tubule promoting sodium retention (which results in water retention and volume expansion with hypertension. There is also excretion of potassium that leads to hypokalaemia.
· Causes:
· a) Primary hyperaldosteronism (independent of the renin-angiotensin axis):
· i) Adrenal adenoma (Conn’s syndrome). 80% of all cases of hyperaldosteronism. The adenomas are usually unilateral and solitary.
· ii) Adrenal Hyperplasia. In bilateral adrenal hyperplasia (BAH) the adrenal cells become hyperplastic, resulting in excessive aldosterone secretion. It accounts for the 15% of all cases of hyperaldosteronism. Unilateral adrenal hyperplasia is rarer and is treated with adrenalectomy.
· iii) Familial hyperaldosteronism. Type 1 is glucocorticoid-remediable aldosteronism (GRA), and type 2 is inherited aldosterone-producing adenoma or an inherited bilateral adrenal hyperplasia. In type 1 the type of inheritance is autosomal dominant and is associated with hypertension at an early age (usually in the ’20s) that it can be resistant to treatment. Patients may develop cerebral aneurysms that can cause hemorrhagic stroke. This subset of patients is normokalaemic (unless treated with diuretics that cause profound hypokalaemia).
· iv) Adrenal carcinoma (rare, usually diagnosed after a biopsy of a surgically removed adrenal adenoma).
· Presentation: hypertension, hypokalaemia (usually < 3.5 mmol/L, however, 70% of the patients may appear normokalaemic), metabolic alkalosis (hypokalemia and metabolic alkalosis are not so prevalent as once thought). Sodium may be normal or at the high end of normal. Patients may also have polyuria & polydipsia (the reduced ability of the kidneys to concentrate urine), weakness (from hypokalaemia), headaches, and lethargy.
· Investigation: Urea & electrolytes (may show hypokalaemia & hypernatremia), spot rennin & aldosterone levels (aldosterone is high, and rennin is low – if rennin is high or normal this virtually excludes the diagnosis of primary hyperaldosteronism), ECG (electrocardiogram; it may show arrhythmias from electrolyte imbalance), CT/MRI (to locate adrenal adenoma and hyperplasia; MRI is more specific). It may be advised that patients should undergo an adrenal venous sampling before surgery, to detect the source of aldosterone.
· Lying & standing aldosterone levels: aldosterone is affected by upright posture, so blood samples are taken with the patient lying down and repeated after being erect for a few hours. In primary hyperaldosteronism due to adrenal hyperplasia plasma aldosterone increases after 4 hours of standing (usually by more than 30%), however, in adrenal adenoma (Conn’s syndrome), there is typically no alteration in rennin/aldosterone levels with posture.
· Salt loading & aldosterone/ renin levels (however this test is less used today): the patient is loaded with salt (high Na+ diet and slow release sodium tablets) for 2 weeks before samples are taken. Aldosterone/rennin, cortisol, and bicarbonate levels are measured. The salt should suppress plasma aldosterone. Failure to suppress it confirms primary hyperaldosteronism.
· Aldosterone/renin ratio (ARR): it is used for screening for primary hyperaldosteronism in patients with hypertension & hypokalaemia or resistant hypertension. If the aldosterone/renin ratio is > 800, then the patients should be investigated with further imaging (CT/MRI) to locate the source (for the increased ratio, the specificity and sensitivity are about 75 – 100%). If aldosterone is > 1 000 with raised aldosterone/renin ratio, then we have about 90% specificity (this is useful especially on Afro – Caribbean patients that tend to have low renin).
· The results of the aldosterone/renin ratio may be affected by antihypertensive medications, e.g., false-positive results occur with beta – blockers, while false (-) occur with diuretics, ACE (angiotensin-converting enzyme) inhibitors, ARBs (angiotensin receptor blockers) and dihydropyridine calcium – channel blockers (‘dipines’ e.g. amlodipine, felodipine, nicardipine, nifedipine, nimodipine). Alpha-blockers do not seem to affect the ratio.
· b) Secondary hyperaldosteronism (with high renin): renal artery stenosis (RAS), coarctation of the aorta, fibromuscular dysplasia, renin-secreting tumors, liver failure, congestive heart failure (CHF), nephrotic syndrome, Gitelman’s syndrome, Bartter’s syndrome, use of diuretics and malignant hypertension. RAS (renal artery stenosis) should be investigated in patients with hypertension & hypokalemia. Urea & electrolytes may show hypokalaemia & renal impairment. Ultrasonography & Doppler/ Triplex of the renal artery may help. MAG3 or DMSA scans will show the function and blood supply of the kidney. A renal arteriogram is the gold standard and also allows angioplasty (if indicated). On secondary hyperaldosteronism renin & aldosterone levels are high.
· Diagnostic algorithm for diagnosing primary aldosteronism:
· Dexamethasone suppression test (DST): a blood test that assesses the adrenal gland function by measuring how the cortisol levels change in response to injection of dexamethasone. It is used to diagnose Cushing syndrome.
· Physiology: dexamethasone is an exogenous steroid that provides negative feedback to the pituitary gland to suppress the secretion of ACTH (adrenocorticotropic hormone).
· Test: there are 2 variations: low dose test(usually 1 – 2 mg) & high dose test (usually 8 mg) of dexamethasone.
· The low dose suppresses cortisol in people with no pathology in endogenous cortisol production.
· The high dose of dexamethasone exerts negative feedback on pituitary ACTH-producing cells, but not on ectopic ACTH – producing cells or adrenal adenoma.
· Interpretation:
· a) Normal: ACTH before dexamethasone administration is normal, and cortisol decreases on low dose dexamethasone test.
· b) Primary hypercortisolism – Cushing’s syndrome (e.g., exogenous administration of glucocorticoids): ACTH before dexamethasone administration is low or undetectable; cortisol is not suppressed by high or low doses.
· c) Ectopic ACTH production (e.g., by small cell lung cancer; if adrenal tumor is not apparent, then a chest & abdominal CT is indicated to rule out a tumour secreting ACTH): ACTH prior to dexamethasone administration is elevated in hundreds; cortisol is not suppressed by high or low dose dexamethasone test.
· d) Cushing’s disease (e.g., a benign pituitary adenoma that secretes ACTH; a pituitary MRI is needed): ACTH, before dexamethasone administration, is normal to elevated, but not in hundreds; cortisol is not suppressed by low dose dexamethasone test but is suppressed by high dose.
· Note: equivocal results should be followed by a CRH (corticotrophin – releasing hormone) stimulation test, with inferior petrosal sinus sampling.
· Cushing’s syndrome, Cushing’s disease, ectopic Cushing’s & pseudo – Cushing’s:
· a) Cushing syndrome (hypercortisolism) is caused most commonly by exogenous administration of glucocorticoids prescribed by a doctor (also called iatrogenic Cushing’s syndrome) for a disease (e.g., for a collagen disease, or asthma, or immunosuppression, e.g., after an organ transplant). It may also be caused by administration of exogenous synthetic ACTH or administration of medroxyprogesterone acetate (as a contraceptive or for HRT (hormone replacement therapy) or for other conditions such as endometriosis). In iatrogenic Cushing’s the adrenal glands atrophy due to lack of stimulation by ACTH, since glucocorticoids downregulate the production of ACTH.
· Endogenous glucocorticoid overproduction or hypercortisolism that is independent of ACTH is usually due to a primary adrenocortical neoplasm (usually an adenoma but rarely a carcinoma).
· Bilateral micronodular hyperplasia and macronodular hyperplasia are rare causes of Cushing syndrome.
· Cushing syndrome is collated with: rapid weight gain; moodiness, irritability or depression; muscle & bone weakness (osteoporosis); memory & attention dysfunction; diabetes mellitus; hypertension; immune suppression; sleep disturbances; menstrual disorders (e.g. amenorrhea) in women; decreased fertility in men; hirsutism; baldness; and hypercholesterolemia.
· b) Cushing’s disease may be pituitary and caused by a benign pituitary adenoma that secretes ACTH (secondary hypercortisolism; 80% of cases of endogenous Cushing syndrome). It may also be induced by CRH secreting tumors that stimulate pituitary ACTH production (tertiary hypercortisolism/ hypercorticism).
· c) Ectopic or paraneoplastic Cushing’s may occur from tumors outside the regular pituitary-adrenal system that produces ACTH, e.g., with small cell lung cancer, oat cell carcinoma, or carcinoid tumor.
· Ectopic corticotropin-releasing hormone (CRH) secretion leading to increased ACTH secretion comprise a scarce group of cases of Cushing syndrome.
· d) Pseudo – Cushing’s syndrome. Elevated levels of total cortisol may be due to estrogen found in oral contraceptive pills that contain estrogen & progesterone. Estrogen can increase cortisol–binding globulin and cause elevation of total cortisol, however total free cortisol (the active hormone), measured by a 24-hour urinary free cortisol, is normal.
· Pseudo – Cushing may also occur in chronic alcoholics, obese, depressed, people with malnutrition, people with physical stress, and patients with: uncontrolled diabetes mellitus, obstructive sleep apnea, anorexia nervosa, and PCOS (polycystic ovarian syndrome). Patients with HIV under HAART treatment, lipodystrophy, and hypertriglyceridemia make a metabolic profile resembling Cushing, however plasma cortisol is not elevated.
· Diagnostic algorithm for Cushing’s syndrome:
· Pheochromocytoma: a neuroendocrine tumor of the medulla of the adrenal glands (originates from chromatin cells), or extra-adrenal chromaffin tissue that failed to involute after birth, and secretes high amounts of catecholamines, especially epinephrine (adrenaline) & to lesser extent norepinephrine (noradrenaline). Extra-adrenal paragangliomas (often described as extra-adrenal pheochromocytomas) are closely related, though less common, tumors that originate in the ganglia of the sympathetic CNS (central nervous system).
· Pheochromocytomas occur most often during young-adult to mid-adult life.
· Signs & symptoms: headache (the most common symptom); tremor; palpitations; weight loss; ileus; skin sensations; flank pain; tachyarrhythmia (elevated heart rate); hypertension [including paroxysmal (sporadic, episodic) high blood pressure, which sometimes can be more difficult to detect; another clue to the presence of pheochromocytoma is orthostatic hypotension; hypertension may be severe or malignant and not well controlled with standard blood pressure medications]; anxiety (may resemble that of a panic attack); diaphoresis (excessive sweating); pallor; localised amyloid deposits; hyperglycaemia. The most common presentation is headache, excessive sweating, and increased heart rate, with the attack subsiding in less than one hour.
· Causes: up to 25% of pheochromocytomas may be familial; may be related to Von Hippel Lindau disease; neurofibromatosis type 1 or familial paraganglioma, depending on the mutation. It may also be a tumor of the MEN syndrome (multiple endocrine neoplasias) type IIA and type IIB (the other component neoplasms of the syndrome include parathyroid adenomas & medullary thyroid cancer; MEN IIA also presents with hyperparathyroidism, while MEN IIB also presents with mucosal neuroma).
· Diagnosis:
· a) By measuring catecholamines & metanephrines (normetanephrine & metanephrine) in plasma or through a 24-hour urine collection (although urine test is slightly less effective than plasma testing, however, it is still considered highly effective in diagnosis). Also, urinary VMA (vanillyl mandelic acid) is elevated in patients with pheochromocytoma. Plasma metanephrine testing have 96% sensitivity & 85% specificity; while 24-hour urinary collection for catecholamines and metanephrines has 87.5% sensitivity & 99.7% specificity.
· Care should be taken to rule out other causes of adrenergic (adrenalin-like) excess like hypoglycemia, stress, exercise, and drugs affecting the catecholamines like stimulants, methyldopa (an antihypertensive medication), dopamine agonists, or ganglion blocking hypertensives. Also, various foodstuffs (e.g., coffee, tea, bananas, chocolate, cocoa, citrus fruits, and vanilla) can also affect the tests.
· b) Chromogranin A (a member of the granin family of neuroendocrine secretory proteins) is elevated in case of pheochromocytoma.
· c) Clonidine suppression test: οne diagnostic test used in the past for pheochromocytoma is to administer clonidine (a centrally-acting alpha-2 agonist usedas antihypertensive). A healthy adrenal medulla will respond to the clonidine suppression test by reducing catecholamine production; the lack of a response is evidence of pheochromocytoma.
· Imaging tests: imaging by CT or T2 MRI of the head, neck, chest & abdomen may help localize a tumor. Tumors can also be located using an MIBG scan (scintigraphy with iodine – 123 marked metaiodobenzylguanidine). The tumor can also be located even finer with PET-CT or PET-MRI with [18F] fluorodopamine or FDOPA.
· Diagnostic algorithm for suspicion of pheochromocytoma:
{kind=link}
No comments:
Post a Comment