
Dr. James Manos (MD)
January 2, 2016
Tips in Hematology
Volume (3)
2nd edition (revised)
CONTENTS
LYMPHOCYTE DISORDERS
Lymphocytosis
Absolute & relative lymphocytosis
Causes of absolute lymphocytosis
Causes of relative lymphocytosis
Causes of inversion (reversal) of the ratio of lymphocytes to neutrophils
Reactive (atypical) lymphocytes
Causes of increased reactive (atypical) lymphocytes in peripheral blood
Infectious mononucleosis
Lymphocytopenia
MONOCYTES
Monocytosis
EOSINOPHILS
Eosinophilia
BASOPHILS
Basophilia
ANEMIAS
Anemia
Anemia – causes
Microcytic anemia – causes
Iron deficiency anemia
Differential diagnosis of iron deficiency anemia and thalassemia
Differential diagnosis of microcytic hypochromic anemias
Lead poisoning
Zinc protoporphyrin (ZPP) & free erythrocyte protoporphyrin (FEP)
Beta-thalassemia
Alpha thalassemia
Sickle cell anemia (homozygous HbS or HbSS)
Normocytic anemia – causes
Anemia of chronic disease
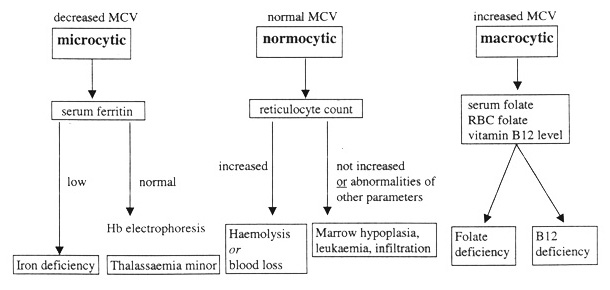
Macrocytic anemia – causes
Megaloblastic anemia
Folate deficiency
Vitamin B12 deficiency
Pernicious anemia
Fanconi anemia
Sideroblastic anemia
Aplastic anemia
Parvovirus B19 & aplastic crisis
Anemia – diagnostic approach (algorithm)
BONE MARROW FAILURE
JAUNDICE/ HYPERBILIRUBINEMIA
Jaundice – overview
Jaundice – table of diagnostic tests
Neonatal jaundice
Hyperbilirubinemia in the term newborn
Unconjugated hyperbilirubinemia – causes
Jaundice – diagnostic approach (algorithm)
HEMOLYSIS/ HEMOLYSIS RELATED DISORDERS
Hemolysis/ hemolytic anemia (overview)
Hemolysis – Workup
Features of hemolysis
Hemolysis - characteristics
Hemolysis – investigation
Intravascular hemolysis
Extravascular hemolysis
HELLP syndrome (on the pregnant)
Hemolytic anemia
Autoimmune hemolytic anemia
Alloimmune hemolytic anemia
Cold agglutinin disease
Direct antiglobulin test (DAT; also called direct Coombs test)
Indirect antiglobulin test (also called indirect Coombs)
Rhesus hemolytic disease of the newborn (Rh HDN)
Hereditary spherocytosis
G6PD deficiency
Idiopathic thrombocytopenic purpura (ITP)
Thrombotic thrombocytopenic purpura (TTP; also known as Moschcowitz syndrome)
Hemolytic uremic syndrome (HUS)
Microangiopathic hemolytic anemia (MAHA)
Mechanical hemolytic anemia
Paroxysmal nocturnal hemoglobinuria (PNH)
Evans syndrome
DIC (disseminated intravascular coagulation)
NEWBORN – HEMATOLOGICAL FEATURES
Newborn – hematological features
LYMPHOCYTE DISORDERS
· Lymphocytosis
· Absolute & relative lymphocytosis:
· In absolute lymphocytosis, the total lymphocyte count is elevated. In adults, absolute lymphocytosis is present when the absolute lymphocyte count is > 4,000/µL; in older children,> 7,000/µL; and in infants,> 9,000/µL.
· Lymphocytes normally represent 20 to 40% of circulating WBCs.
· Relative lymphocytosis occurs when there is a higher proportion (> 40%) of lymphocytes among the WBCs, while the absolute lymphocyte count (ALC) is normal (< 4,000 / microliter). Relative lymphocytosis is normal in children under age 2.
· Causes of absolute lymphocytosis: Causes of absolute lymphocytosis include acute viral infections [such as infectious mononucleosis (the majority from EBV virus; also may be caused by CMV virus), HIV, hepatitis, CMV, etc.), other acute infections such as pertussis (causes whooping cough); some protozoal infections [such as toxoplasmosis and Chagas disease (American trypanosomiasis)], chronic intracellular bacterial infections [such as tuberculosis (TB) and brucellosis], CLL (chronic lymphocytic leukemia), ALL (acute lymphoblastic leukemia), lymphoma, post-splenectomy, etc.
· Other causes of lymphocytosis include autoimmune/ connective tissue (collagen) diseases, drugs, thyrotoxicosis, Addison’s disease, splenomegaly, and post-vaccination.
· Causes of relative lymphocytosis: acute viral infections, connective tissue diseases, thyrotoxicosis, Addison’s disease, and splenomegaly with splenic sequestration of granulocytes. It may occur at an age under 2 years.
· Inversion (reversal) of the ratio of lymphocytes to neutrophils: inversion (‘reversal’) of the ratio of lymphocytes to neutrophils is a mark of some types of viral infection or may indicate other diseases.
· Causes of inversion (reversal) of the ratio of lymphocytes to neutrophils:
· a) Viral infection. Mild neutropenia and lymphocytosis are common manifestations of several viral infections, including mumps, measles, influenza, hepatitis, and infectious mononucleosis (the majority due to EBV; less so with CMV). High numbers of atypical lymphocytes contribute to the decrease in neutrophils by affecting neutrophil production and maturation processes.
· b) Adult-onset cyclic neutropenia. Cyclic neutropenia is a genetic disorder characterized by periodic decreases in neutrophil count. During the neutropenic phase, which lasts for 3 – 5 days, the number of neutrophils (ANC) drops below 200 cells/ µl. After this phase, the counts increase and remain at 200 cells per µl of blood for about 21 days. Such a decrease results from a genetic mutation that shortens neutrophil lifespan. The adult-onset form of this disease is characterized by lymphocytosis due to an increase in large granular lymphocytes (LGLs). The patient often experiences repetitive episodes of malaise, fever, infections, and ulcers. Treatment includes administration of granulocyte colony-stimulating factor (G-CSF). Symptomatic treatments include the use of steroids and antibiotics such as cephalosporins.
· c) Lymphoma. Lymphomas are characterized by the formation of solid tumors resulting from an abnormal accumulation of mature lymphocytes. They are broadly categorized as Hodgkin's and non-Hodgkin's lymphomas. When such tumorigenic cells infiltrate the bone marrow, neutrophil proliferation and differentiation are impaired, leading to neutropenia, as reflected by a low absolute neutrophil count (ANC). Common symptoms include swollen lymph nodes, fever, night sweats, fatigue, itchy skin, and abdominal pain and discomfort due to splenomegaly and/or hepatomegaly.
· d) Leukemia. The several types of leukemias associated with neutropenia include large granular lymphocyte (LGL) leukemia, T-cell large granular lymphocyte (T-LGL) leukemia, Acute myeloid leukemia (AML), Acute lymphocytic leukemia (ALL), and chronic lymphocytic leukemia (CLL). LGL and T-LGL leukemias arise from uncontrolled proliferation of NK cells and cytotoxic T cells, respectively, and are the most common leukemias that cause neutropenia. AML, ALL, and CLL involve the clonal expansion of progenitor lymphocytes. As a result, the lymphocyte counts in such patients are high. Such uncontrolled clonal expansion also interferes with the normal production and differentiation of neutrophils.
· e) Autoimmune diseases. Certain autoimmune diseases, including Felty syndrome (in association with rheumatoid arthritis; characterized by neutropenia & splenomegaly) and systemic lupus erythematosus (SLE), are inflammatory and involve an increase in the number of LGLs. These cells infiltrate the bone marrow and hamper neutrophil production; thus, a mixed state of neutropenia and lymphocytosis may be observed. The treatments are often symptomatic. Immunosuppressive drugs are also used to reduce immune activity.
· Reactive (atypical) lymphocytes: lymphocytes that become large as a result of Ag (antigen) stimulation; typically, they can be more than 30 μm in diameter with varying size and shape. The nucleus can be round, elliptic, indented, cleft, or folded. The cytoplasm is often abundant and can be basophilic. Vacuoles and/or azurophilic granules can also sometimes be present. Most often, the cytoplasm is gray, pale blue, or deep blue. The distinctive cell associated with EBV or CMV is known as a ‘Downey cell.’ Reactive (atypical) lymphocytes appear larger with more abundant cytoplasm, oval or irregular nucleus that may contain a nucleolus, and show peripheral basophilic cytoplasm and scalloping of borders around red cells.
· Causes of increased reactive (atypical) lymphocytes in peripheral blood: infections from Epstein-Barr virus (EBV), cytomegalovirus (CMV) that cause infectious mononucleosis (CMV causes the minority of the cases); infections from rubella, roseola, syphilis, toxoplasmosis, and viral hepatitis (e.g., hepatitis C), group B streptococci (GBS, Streptococcus agalactiae), Hantavirus; other causes include drugs (such as phenytoin), radiation, hormonal causes (e.g., Addison’s disease), autoimmune diseases (e.g., collagen diseases such as rheumatoid arthritis (RA)); vaccination, and lymphoproliferative disorders (e.g., common variable immune deficiency, Chediak-Higashi syndrome, Wiskott-Aldrich syndrome, X-linked lymphoproliferative disorders).
· Infectious mononucleosis (glandular fever; also called ‘kissing disease’ (as it may be transmitted with a kiss)): caused in 90% of cases by EBV (Epstein–Barr virus) but may also be caused by CMV (Cytomegalovirus). It is more common in adolescents and young adults.
· Signs & symptoms in infectious mononucleosis: fever, sore throat/ pharyngitis, fatigue, lymphadenopathy (usually the posterior cervical LNs (lymph nodes), splenomegaly, petechiae on the roof of the mouth, and, uncommonly, a palatal enanthem. Hepatomegaly may also occur. A maculopapular rash usually develops if an ampicillin or amoxicillin antibiotic is given. It is self–limiting.
· Peripheral blood in infectious mononucleosis: atypical (reactive) lymphocytosis. Lymphocytes > 50% of cells on differential leukocyte count; >_ 10% of the lymphocytes are atypical (see below).
· Lab tests: heterophil Abs (Monospot test - older test: Paul Bunnell) and EBV-specific Abs (antibodies) in serum.
· Lymphocytopenia (also called lymphopenia): an abnormally low level of lymphocytes in the peripheral blood.
· Classification:
· a) T lymphocytopenia: there are too few T lymphocytes (T cell deficiency) but normal numbers of other lymphocytes. It is usually caused by HIV infection. It may also be caused by idiopathic CD4+ lymphocytopenia (ICL), a very rare, heterogeneous disorder characterized by CD4+ T-cell counts below 300 cells/µL in the absence of any known immune deficiency condition, such as human immunodeficiency virus (HIV) infection or chemotherapy.
· b) B lymphocytopenia: there are too few B lymphocytes, but possibly normal numbers of other lymphocytes. It presents as a humoral immune deficiency and is usually caused by medications that suppress the immune system.
· c) NK lymphocytopenia: there are too few ΝΚ (natural killer) cells but normal numbers of other lymphocytes. It is very rare.
· Causes οf lymphocytopenia: recent infection, such as common cold (recent infection is the most common cause of temporary lymphocytopenia), corticosteroids, infections (bacterial, viral (especially HIV), fungal), malnutrition, autoimmune/collagen diseases [including SLE (systemic lupus erythematosus), rheumatoid arthritis (RA), sarcoidosis, etc.), severe stress, intense or prolonged physical exercise (due to cortisol release), iatrogenic (such as chemotherapy e.g. with cytotoxic or immunosuppressive medications for malignancy or autoimmune diseases), radiation exposure/ radiotherapy (high doses), and blood malignancies, such as leukemia and advanced Hodgkin’s disease.
MONOCYTES
· Monocytosis: an increase in the absolute count of monocytes in the peripheral blood. Monocytes are white blood cells that give rise to macrophages and dendritic cells in the immune system.
· Causes of monocytosis: chronic inflammation such as infections [e.g., tuberculosis (TB), brucellosis, listeriosis, subacute bacterial endocarditis (SBE), syphilis, rickettsial infections (e.g., Rocky Mountain spotted fever caused by Rickettsia rickettsii), malaria, kala-azar (visceral leishmaniasis), viral infections, protozoal infections, etc.); blood malignancies (e.g., myeloproliferative disorders; Hodgkin’s lymphoma, certain leukemias such as chronic myelomonocytic leukemia (CMML) and monocytic leukemia), autoimmune diseases (e.g., systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), inflammatory bowel disease (IBD), sarcoidosis, etc.); immune causes (e.g., chronic neutropenia), the recovery phase of neutropenia or an acute infection; obesity, lipid storage diseases, etc.
EOSINOPHILS
· Eosinophilia: the increase in the absolute eosinophil count in the peripheral blood > 4.5 x 108/L (450/μl). Causes: allergic diseases (including allergic asthma, urticaria, allergic rhinitis (hay fever), drug reactions), parasitic infection (filaria, trichinosis, toxocariasis, strongyloidiasis, echinococcosis; eosinophilia is associated with helminth infections, but even then it may be absent), dermatological disorders (eczema, dermatitis herpetiformis (associated with gluten intolerance), bullous pemphigoid), carcinoma after radiotherapy, pulmonary disorders (e.g. pulmonary eosinophilia e.g. Loffler’s syndrome (associated with parasite infection such as Ascaris lumbricoides), tropical eosinophilia); hematological malignancies (myeloproliferative disorders such as CML (chronic myelogenous leukemia), mastocytosis and chronic eosinophilic leukemia; ALL (acute lymphoblastic leukemia); Hodgkin’s lymphoma; some forms of non – Hodgkin lymphoma; acute eosinophilic leukemia; peripheral T – cell lymphoma), idiopathic hypereosinophilic syndrome, eosinophilic esophagitis or gastroenteritis, systemic autoimmune diseases (e.g. SLE (systemic lupus erythematosous), idiopathic eosinophilic synovitis, some forms of vasculitis (e.g. Churg – Strauss syndrome), cholesterol embolism (transiently), coccidiomycosis, HIV infection, interstitial nephropathy, hyperimmunoglobulin E syndrome etc.
· Note: eosinophilic granulomatosis with polyangiitis (also known as Churg–Strauss syndrome or allergic granulomatosiς) is an autoimmune condition that causes inflammation of small and medium-sized blood vessels (vasculitis) in persons with a history of airway allergic hypersensitivity (atopy). It usually manifests in three stages: a) Early (prodromal) stage, characterized by airway inflammation; almost all patients experience asthma and/or allergic rhinitis. B) Second stage: characterized by eosinophilia, which causes tissue damage, most commonly to the lungs and the digestive tract. c) The third stage consists of vasculitis, which can eventually lead to cell death and can be life-threatening.
BASOPHILS
· Basophilia: increased absolute count of basophils. Causes: allergy or inflammation [ulcerative colitis (UC), drug/food hypersensitivity (e.g., nuts, seafood)/inhalant (e.g., pollen) erythroderma, urticaria, juvenile rheumatoid arthritis], endocrinopathy [diabetes mellitus (DM), estrogens, hypothyroidism], infection (TB (tuberculosis), influenza, chickenpox, smallpox), iron deficiency, ionizing radiation, basophilic leukemia, carcinoma, myeloproliferative neoplasms (especially CML (chronic myelogenous leukemia), polycythemia vera, primary myelofibrosis, essential thrombocythemia).
ANEMIA
· Anemia: Anemia is strictly defined as a decrease in red blood cell (RBC) mass. The function of the RBC is to deliver oxygen from the lungs to the tissues and carbon dioxide from the tissues to the lungs. This is accomplished by using hemoglobin (Hb), a tetramer protein composed of heme and globin. Anemia impairs the body’s ability for gas exchange by decreasing the number of RBCs transporting oxygen (O2) and carbon dioxide (CO2). Anemia, like a fever, is a sign that requires investigation to determine the underlying etiology. Often, practicing physicians overlook mild anemia. Anemia is considered to be present if the Hb or the hematocrit (Hct) is below the lower limit of 2 standard deviations (-2SD) or the 95% confidence interval for the normal population. This definition of anemia results in 2.5% of normal individuals being classified as anemic. Anemia is absolute if the RBC mass is decreased, and relative if it is associated with increased plasma volume.
· Absolute anemia can be divided into 2 main categories: decreased RBC production and elevated RBC destruction or loss more than the bone marrow’s ability to replace those losses.
· Causes of decreased RBC production include nutritional deficiencies (iron, folate, vitamin B12, vitamin B6), anemia of chronic disease, renal, liver, or endocrine disease, bone marrow infiltration (myelophthisic anemia), aplastic anemia, pure red cell aplasia, and sideroblastic anemia.
· Causes of increased RBC destruction or loss more than the bone marrow’s ability to replace those losses include blood loss (hemorrhage), hemolysis of various etiologies (both intrinsic and extrinsic), and hemoglobin disorders (hemoglobinopathies and thalassemias).
· Relative anemia may be seen in overhydration (volume overload), pregnancy, macroglobulinemia, and post-flight astronauts.
· Anemia – causes: basically, only three causes of anemia exist: blood loss, increased destruction of RBCs (hemolysis), and decreased production of RBCs. Each of these causes includes some disorders that require specific and appropriate therapy.
· Genetic etiologies include:
· a) Hemoglobinopathies.
· b) Thalassemias.
· c) Enzyme abnormalities of the glycolytic pathways.
· d) Defects of the RBC cytoskeleton.
· e) Congenital dyserythropoietic anemia.
· f) Rh null disease (lack of all Rh antigens; stomatocytosis & hemolysis)
· g) Hereditary xerocytosis.
· h) Abetalipoproteinemia.
· i) Fanconi anemia.
· Nutritional etiologies include:
· a) Iron deficiency.
· b) Vitamin B-12 deficiency.
· c) Folate deficiency.
· d) Starvation and generalized malnutrition.
· Physical etiologies include:
· a) Trauma.
· b) Burns.
· c) Frostbite.
· d) Prosthetic valves and surfaces.
· Chronic disease and malignant etiologies include:
· a) Renal disease.
· b) Hepatic disease.
· c) Chronic infections.
· d) Neoplasia.
· e) Collagen vascular diseases.
· Infectious etiologies include:
· a) Viral causes: Hepatitis, infectious mononucleosis, cytomegalovirus (EBV).
· b) Bacterial causes: Clostridia, gram-negative sepsis.
· c) Protozoal causes: Malaria, leishmaniasis, toxoplasmosis.
· Thrombotic thrombocytopenic purpura (TTP) and hemolytic-uremic syndrome may be a cause of anemia.
· Hereditary spherocytosis may present as severe hemolytic anemia or asymptomatic with compensated hemolysis.
· G-6-PD deficiency may manifest as chronic hemolytic anemia or exist without anemia until the patient receives an oxidant medication.
· Immunologic etiologies for anemia may include antibody-mediated abnormalities.
· Acute hemorrhage is, in the emergency department (ED), by far the most common etiology for anemia.
· Drugs or chemicals commonly cause the aplastic and hypoplastic group of disorders. Certain types of these causative agents are dose-related, and others are idiosyncratic. Any human exposed to a sufficient dose of inorganic arsenic, benzene, radiation, or the usual chemotherapeutic agents used for the treatment of neoplastic diseases develops bone marrow depression with pancytopenia. Among idiosyncratic agents, only occasional human exposure to these drugs results in an untoward reaction that suppresses one or more of the formed elements of the bone marrow (1:100 to 1:1,000,000). With certain types of these drugs, pancytopenia is more common, whereas with others, suppression of one cell line is usually observed. Thus, chloramphenicol may produce pancytopenia, whereas granulocytopenia is more frequently found with toxicity to sulfonamides or antithyroid drugs. The idiosyncratic causes of bone marrow suppression include multiple medications, such as antibiotics, antimicrobials, anticonvulsants, and antihistamines.
· Other idiosyncratic causes of known etiology are viral hepatitis and paroxysmal nocturnal hemoglobinuria. In approximately 50% of patients with aplastic anemia, a definite etiology cannot be established, and the anemia must be considered idiopathic.
· Rare causes of anemia due to hypoplastic bone marrow include familial disorders and the acquired pure red cell aplasias characterized by a virtual absence of erythroid precursors in the bone marrow, with normal numbers of granulocytic precursors and megakaryocytes.
· Rare causes of diminished erythrocyte production with hyperplastic bone marrow include hereditary (inherited) orotic aminoaciduria and erythremic myelosis.
· Microcytic anemia – causes iron deficiency anemia, thalassemias, sideroblastic anemia, anemia of chronic disease.
· Normocytic anemia – causes:
· a) Normal reticulocytes: recent blood loss; hemolytic anemia.
· b) Deficient reticulocyte production: aplastic anemia.
· c) Myelophthisic anemia: chronic renal failure, anemia of chronic disease, hypothyroidism.
· Iron deficiency anemia: Has decreased MCV, RBC count, serum iron, transferrin saturation, and serum ferritin. Low or absent marrow hemosiderin. Has increased RDW (in >90% of patients).
· RBCs microcytic & hypochromic, pencil cells, anisocytosis; increased TIBC, FEP (free erythrocyte protoporphyrin) & S. TfR (serum transferrin receptor).
· Normal Hb electrophoresis.
· In iron deficiency anemia, platelets may be increased.
· Resistant iron deficiency anemia may indicate malabsorption, e.g., coeliac disease.
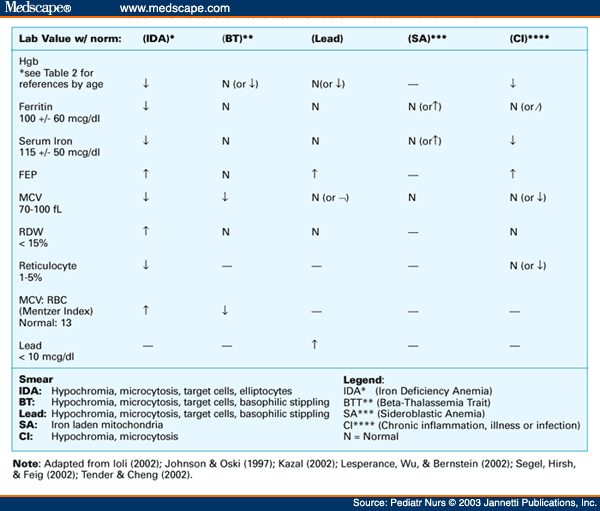
· Differential diagnosis of iron deficiency anemia, anemia of chronic disease, and thalassemia:
· The mean corpuscular volume (MCV), red blood cell (RBC) distribution width (RDW), and the patient's history can exclude some of these etiologies.
· The MCV is usually < 75 fL in thalassemia and rarely < 80 fL in iron deficiency until the hematocrit is < 30%.
· Elevated RBC count with markedly decreased MCV differentiates thalassemia from iron deficiency anemia.
· The Mentzer index (MCV/RBC count) can help distinguish between iron deficiency and thalassemia. The index is usually >13 in iron deficiency and <13 in thalassemia. A ratio of 13 would be considered uncertain. In iron deficiency, the marrow cannot produce as many RBCs, and they are small (microcytic), so the RBC count and the MCV will both be low, and as a result, the index will be greater than thirteen.
Conversely, in thalassemia, which is a disorder of globin synthesis, the number of RBCs produced is normal, but the cells are smaller and more fragile. Therefore, the RBC count is normal, but the MCV is low, so the index will be less than 13. Limitations: In practice, the MCV must be extremely low and/or the RBC extremely high to produce an index of less than 13, which is rarely encountered, further calling into question the index's usefulness.
Conversely, in thalassemia, which is a disorder of globin synthesis, the number of RBCs produced is normal, but the cells are smaller and more fragile. Therefore, the RBC count is normal, but the MCV is low, so the index will be less than 13. Limitations: In practice, the MCV must be extremely low and/or the RBC extremely high to produce an index of less than 13, which is rarely encountered, further calling into question the index's usefulness.
· A retrospective study on differentiating between beta-thalassemia trait (β-TT) and iron deficiency anemia (IDA) concluded that the Mentzer index was the most reliable index, as it had the highest sensitivity (98.7%), specificity (82.3%), and Youden’s index (81%) for detecting β-TT; this was followed by the Ehsani index (94.8%, 73.5%, and 68.3%, resp.) and RBC count (94.8%, 70.5%, and 65.3%). The study concluded that the Mentzer index had the highest reliability in differentiating β-TT from IDA (Reference: http://www.hindawi.com/journals/anemia/2014/576738/).
· RDW can help differentiate iron deficiency and sideroblastic anemia from thalassemia. RDW is elevated in > 90% of individuals with iron deficiency, but in only 50% of patients with thalassemia. RDW is usually elevated in sideroblastic anemia. Thalassemia will almost always be the cause of microcytic anemia with a normal RDW, but individuals with an elevated RDW require additional testing.
· Serum ferritin is the best test to screen for iron deficiency anemia. In the absence of inflammation, a normal serum ferritin level generally excludes iron deficiency.
· Sideroblastic anemia can be excluded with a peripheral blood smear or bone marrow aspirate.
· A normal serum lead level excludes lead poisoning.
· Anemia associated with chronic disease is most often a normocytic, normochromic, mild anemia. If thalassemia is still suspected, hemoglobin electrophoresis may help the diagnosis.
· Reticulocyte count in thalassemia is usually elevated. The degree of elevation depends upon the severity of thalassemia. Chronic extravascular hemolysis occurs because of ineffective erythropoiesis, so indirect bilirubin is elevated in thalassemia major and intermedia.
· On iron deficiency anemia, TIBC increases, and serum ferritin decreases. On thalassemia, TIBC and serum ferritin are normal.
· Free erythrocyte porphyrin (FEP) is normal in thalassemia; increased in anemia of chronic disease, iron deficiency, and lead poisoning; and decreased in sideroblastic anemia.
· Lead poisoning: findings may include basophilic stippling of the RBCs on peripheral blood smears; basophilic stippling is not specific for lead toxicity and may be observed in arsenic toxicity, sideroblastic anemia, and thalassemia. The anemia of lead toxicity may be normocytic or microcytic. A classic presentation of chronic metal exposure includes anemia, Mees lines (horizontal hypopigmented lines across all nails), and subtle neurologic findings. Patients with chronic metal toxicity tend to have more prominent central and peripheral nervous system involvement. However, encephalopathy and peripheral neuropathies may occur within a few hours to days of acute high-dose exposure. A blue line and gum with bluish-black edging to the teeth, known as a Burton line, indicate chronic lead poisoning. Children with chronic poisoning may refuse to play or may have hyperkinetic or aggressive behavior disorders. Visual disturbance may present with gradually progressing blurred vision because of central scotoma caused by toxic optic neuritis.
· Zinc protoporphyrin (ZPP) & free erythrocyte protoporphyrin (FEP):
· Zinc protoporphyrin (ZPP) is primarily ordered to help detect iron deficiency in children and to detect and monitor chronic exposure to lead in adults. ZPP is a substance that is typically found in small amounts in RBCs. Most of the protoporphyrin in red blood cells combines with iron to form heme, the molecule in hemoglobin that carries oxygen. Zinc combines with protoporphyrin instead of iron when there is insufficient iron available to form heme, as in iron deficiency, or when lead is present and blocks the formation of heme, as in lead poisoning. The ZPP level in the blood will rise under these conditions. Two types of tests are available to measure ZPP: a) The free erythrocyte protoporphyrin (FEP) test measures both ZPP, which accounts for 90% of protoporphyrin in red blood cells, and free protoporphyrin, which is not bound to zinc. b) The ZPP/heme ratio gives the proportion of ZPP compared to the normal iron-containing heme in red blood cells.
· Free erythrocyte porphyrin (FEP) is normal in thalassemia; increased in anemia of chronic disease, iron deficiency, and lead poisoning; and decreased in sideroblastic anemia.
· Differential diagnosis of microcytic hypochromic anemias:
· RDW: increased in iron deficiency & sideroblastic anemia but remains normal in thalassemia, lead poisoning, and anemia of chronic diseases.
· Serum iron: decreased in iron deficiency and in anemia of chronic diseases but increased in sideroblastic anemia while remaining normal in lead poisoning and thalassemia.
· TIBC: increased in iron deficiency but remains normal in lead poisoning, sideroblastic anemia, and thalassemia, while decreased in anemia of chronic diseases.
· Serum ferritin: decreased in iron deficiency but increased in sideroblastic anemia and anemia of chronic diseases (ferritin is an inflammatory acute-phase protein) while remaining normal in lead poisoning and thalassemia.
· Free erythrocyte porphyrin (FEP): normal in thalassemia but increased in anemia of chronic disease, iron deficiency, and lead poisoning, while decreased in sideroblastic anemia.
· Beta – thalassemia:
· Classification:
· a) Major: βο/βο
· b) Intermedia: β+/β+ or βο/β+
· c) Minor: β+/β or βοβ.
· Laboratory features: Markedly decreased MCV; normal RBC count; normal serum iron and TIBC; normal or increased transferrin saturation; normal serum ferritin; normal marrow hemosiderin. Elevated or normal RDW.
· Blood film: RBCs microcytic hypochromic, basophilic stippling, target cells, polychromasia; normal TIBC and FEP (free erythrocyte protoporphyrin); increased S. TfR (serum transferrin receptor).
· Hb electrophoresis: HbA2 > 3.5%; hemoglobin is mainly HbF with absent HbA in βο/βο thalassemia; some amount of HbA is present in βοβ+ and β+β+ thalassemia.
· Blood smear: anisopoikilocytosis, hypochromia, microcytosis, basophilic stippling, target cells, nucleated RBCs.
· MCV is reduced. Reticulocytosis is slight due to ineffective erythropoiesis.
· Alpha thalassemia:
· Classification:
· a) Hydrops fetalis (stillbirth newborn): -/- -/- (Barts hemoglobin).
· b) Hemoglobin H disease: -/- -/α (microcytic hypochromic anemia, blood smear: target cells, Heinz bodies, hepatosplenomegaly).
· c) alpha – thalassemia minor: -/- α/α/ or -/α -/α (mild microcytic hypochromic anemia).
· d) Thalassemia minima: -/α α/α (silent carriers; may have reduced MCV & MCH).
· Sickle cell anemia (homozygous HbS or HbSS):
· Lab features: variable anemia, a positive sickling test, and solubility test, Hb electrophoresis & HPLC (high-performance liquid chromatography) show HbS as the major hemoglobin and the remaining being HbF.
· Common manifestations: hemolytic anemia, hand & foot syndrome, crisis, chest syndrome, gallstones, chronic leg ulcers.
· Complications: infections, vaso-occlusive crises, chest syndrome, renal infarcts & papillary necrosis, kidney failure, avascular necrosis of the femoral head, osteomyelitis from Salmonella.
· Anemia of chronic disease (ACD): normal or decreased MCV; decreased RBC count and serum iron; decreased or normal transferrin saturation; normal or increased serum ferritin; normal or increased marrow hemosiderin. Normal or increased RDW.
· RBCs normocytic normochromic or microcytic hypochromic; decreased TIBC; increased FEP (free erythrocyte protoporphyrin); normal S. TfR (serum transferrin receptor); normal Hb electrophoresis.
· Causes of anemia of chronic disease: chronic infections [including TB (tuberculosis), UTIs (urinary tract infections), deep-seated abscess, pneumonia, subacute endocarditis) etc.], chronic inflammation (e.g., collagen diseases), and neoplasms (solid/ blood malignancies).
· Macrocytic anemia – causes megaloblastic anemia (B12 and/or folate deficiency, pernicious anemia), liver disease, hemolytic anemia, alcoholism, MDS (myelodysplastic disorder), hypothyroidism, chemotherapy (such as hydroxyurea) and antifolates (such as phenytoin) and bone marrow infiltration.
· Megaloblastic anemia: pancytopenia; triad (on peripheral blood smear): oval macrocytes + hypersegmented PMNs (> 5 nuclear lobes; > 5%) + Howell Jolly bodies. In severe cases, you may see Cabot rings, basophilic stippling, Howell-Jolly, & pancytopenia. The reticular count is low for the degree of anemia.
· Causes: B12 and/or folate deficiency; pernicious anemia, MDS (myelodysplastic syndrome), HIV, inherited pyrimidine synthesis disorders (such as orotic aciduria), inherited DNA synthesis disorders, erythroleukemia, alcohol abuse, hypothyroidism, and toxins and drugs such as folic acid antagonists (such as methotrexate, trimethoprim, pentamidine), purine synthesis antagonists (such as 6-mercaptopurine), pyrimidine antagonists (such as cytosine arabinoside); phenytoin (for epilepsy), nitrous oxide (for anesthesia), hydroxyurea etc.
· Folate deficiency: serum folate & RBC folate are low, and urine FIGlu is increased. Homocysteine levels are elevated (this may also occur in vitamin B12 deficiency), but contrary to vitamin B12 deficiency, methylmalonic acid (MMA) is normal.
· Causes: insufficient dietary (low diet of green leafy vegetables; the commoner cause), chronic alcoholism, malabsorption (e.g. celiac disease), pregnancy, hemolytic anemias, neoplasms, drugs - including antifolates (e.g. methotrexate (MTX), trimethoprim, pentamidine), liver cirrhosis, pregnancy, infants, rapid cellular proliferation, surgery (intestinal and/or jejunal resection), nutritional deficiency of thiamine and factors such as enzymes responsible for folate metabolism etc.
· Vitamin B12 deficiency: Serum methylmalonic acid is increased. Serum vitamin B12 is low. Homocysteine levels are elevated (this may also occur in vitamin B12 deficiency), but in folate deficiency, unlike in vitamin B12 deficiency, methylmalonic acid (MMA) is normal. Schilling test. Vitamin B12 deficiency may also cause neurological disease (not caused by pure folate deficiency).
· Causes of vitamin B12 deficiency: strict vegetarians/ vegans (vegans are like vegetarians but do not eat dairy products), pernicious anemia, coeliac disease, tapeworm infestation (especially the marine parasite (fish tapeworm) Diphyllobothrium spp.), gastrectomy, achlorhydria (stomach), chronic pancreatitis, surgery (iliac resection & bypass), blind loop syndrome (intestinal surgery; we have bacterial overgrowth), biological competition for vitamin B12 by diverticulosis, fistula, intestinal anastomosis), congenital (juvenile megaloblastic anemia), drugs, nitrous oxide (NO) anesthesia (usually requires repeated instances), etc.
· The most frequent cause of vitamin B12 deficiency in people who are not vegans is deficient absorption.
· Pernicious anemia: morphological features of megaloblastic anemia in blood & marrow, low serum vitamin B12, abnormal Schilling test (radiolabeled vitamin B12 is given orally), pentagastrin-fast achlorhydria (chronic atrophic gastritis), absence of hydrochloric acid in gastric juice, anti – IF (intrinsic factor – it is produced by parietal cells of gastric mucosa) & parietal cell Abs (antibodies) in serum. Pernicious anemia is one of many types of megaloblastic anemias.
· It may be caused by the loss of gastric parietal cells, which are responsible, in part, for the secretion of intrinsic factor (a protein essential for subsequent absorption of vitamin B12 in the ileum). A common underlying cause is atrophic gastritis with autoimmune destruction of gastric parietal cells (and autoantibody inactivation of intrinsic factor), leading to a lack of intrinsic factor (vitamin B12 absorption in the gut is normally dependent on intrinsic factor, and its loss leads to vitamin B12 deficiency).
Antibodies to gastric parietal cells and intrinsic factors are prevalent in PA. Parietal cell antibodies are found in other autoimmune disorders and in up to 10% of healthy individuals, making the test nonspecific. However, around 85% of PA patients have parietal cell antibodies, making them a sensitive marker for the disease, since only 50% of individuals in the general population with these antibodies have pernicious anemia. Intrinsic factor antibodies are much less sensitive than parietal cell antibodies but are much more specific. They are found in about half of PA patients and are very rarely seen in other disorders. These antibody tests can distinguish between PA and dietary B12 malabsorption. It has been suggested that combining tests for intrinsic factor and parietal cell antibodies could improve the overall sensitivity and specificity of the diagnostic results. Αtrophic gastritis type A diagnosis should be confirmed by gastroscopy and biopsy.
· Fanconi anemia (FA): It should not be confused with Fanconi syndrome, a kidney disorder. Fanconi anemia is a genetic disease with an incidence of 1: 350000 births, with a higher frequency in Ashkenazi Jews (the carrier frequency in the Ashkenazi Jewish population is about 1/90) and Afrikaners in South Africa.
· It is primarily an autosomal recessive genetic disorder. There is a 25% risk that each subsequent child will have FA. About 2% of FA cases are X-linked recessive, which means that if the mother carries one mutated Fanconi anemia allele on one X chromosome, there is a 50% chance that male offspring will present with Fanconi anemia. Scientists have identified 17 FA or FA-like genes.
· FA is the result of a genetic defect in a cluster of proteins responsible for DNA repair. As a result, most FA patients develop cancer, most often acute myelogenous leukemia (AML), and 90% develop bone marrow failure by age 40. About 60–75 percent of FA patients have congenital defects, commonly short stature, abnormalities of the skin, arms (such as radial deficiencies), head, eyes, kidneys, and ears, and developmental disabilities.
· Around 75% of FA patients have some form of endocrine problem, with varying degrees of severity. The median age of death was 30 years in 2000. Short stature and skin pigmentation may become apparent during childhood, with café au lait spots among the features. The first signs of a hematologic problem are usually petechiae and bruises, followed by paleness & tiredness (anemia) and infections.
· Because macrocytosis usually precedes thrombocytopenia, patients with congenital anomalies associated with FA should be evaluated for an elevated MCV.
· Treatment with androgens and hematopoietic (blood cell) growth factors can temporarily help treat bone marrow failure, but the long-term treatment is with a bone marrow transplant. Cells from people with FA are sensitive to DNA crosslinking drugs used to treat cancer, e.g., mitomycin C.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Sideroblastic anemia: Anemia, ringed sideroblasts in bone marrow, ineffective erythropoiesis.
· Mitochondrial defect.
· Causes of sideroblastic anemia: hereditary or acquired MDS (myelodysplastic syndrome), AML (acute myeloid leukemia), alcoholism, lead poisoning, drugs (e.g., isoniazid and chloramphenicol).
Aplastic anemia: Pancytopenia, decreased cellularity in bone marrow.
· Lab features anemia, neutropenia, thrombocytopenia, and reticulocyte production index < 2.
· Causes of aplastic anemia:
· a) Acquired: idiopathic, drugs [antibiotics such as chloramphenicol; antiepileptics such as carbamazepine, felbamate, and phenytoin); quinine; phenylbutazone (NSAID for animals)], chemicals, ionizing radiation, infections (e.g., viral hepatitis, CMV, EBV), paroxysmal nocturnal hemoglobinuria, GvHD (graft versus host disease), pregnancy.
· b) Constitutional: Fanconi anemia, dyskeratosis congenita, syndrome Schwachman – Diamond.
· Parvovirus B19 & aplastic crisis: parvovirus infection may cause decreased erythropoiesis, called aplastic crisis (or reticulocytopenia), that is most dangerous on patients with pre-existing bone marrow stress such as sickle cell anemia or hereditary spherocytosis.
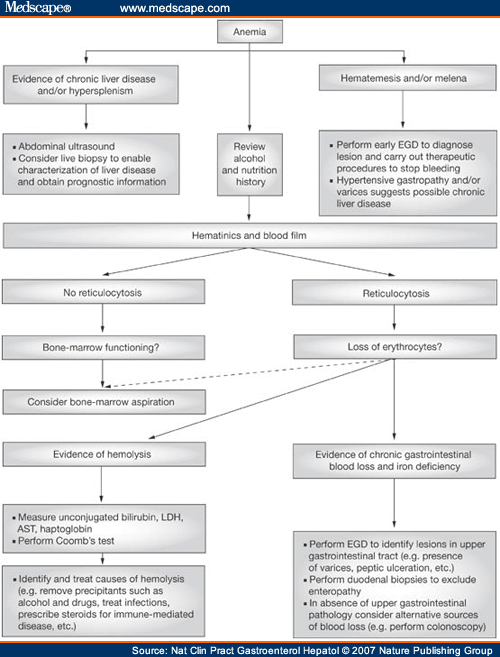
· Anemia – diagnostic approach (algorithm):
· Anemia (generally):
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Anemia – MCV based algorithm:
{kind=link}
· Anemia in the elderly:
{kind=link}
· Macrocytic Anemia in the elderly:
{kind=link}
· Chronic anemia:
{kind=link}
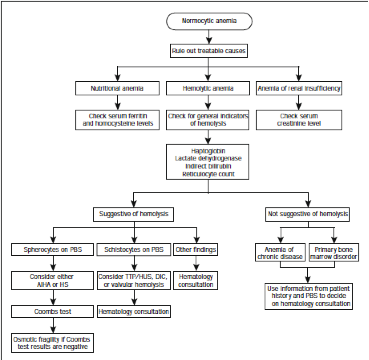
· Hemolytic anemia:
{kind=link}
· Macrocytic Anemia:
{kind=link}
· Microcytic Anemia:
{kind=link}
{kind=link}
{kind=link}
· Normocytic Anemia:
{kind=link}
{kind=link}
{kind=link}
· Anemia in alcoholics:
{kind=link}
· Anemia in children:
{kind=link}
· Normocytic Anemia in children:
{kind=link}
· Macrocytic Anemia in children:
{kind=link}
· Microcytic Anemia in children:
{kind=link}
· The bone marrow failure syndromes include a group of disorders that can be either inherited or acquired. These diseases are disorders of the hematopoietic stem cell that can involve either 1 cell line or all the cell lines (erythroid for red cells, myeloid for white blood cells, megakaryocytic for platelets). The lymphocytes, which are involved in lymphoproliferative disorders, are usually spared.
· Prevalence: the prevalence of bone marrow failure resulting from hypoplastic or aplastic anemia is low in the United States and Europe (2 – 6 cases per million persons) compared with the prevalence of bone marrow failure resulting from acute myelogenous leukemia and multiple myeloma (27 – 35 cases per million persons).
· Bone marrow failure can be inherited or acquired, involving a single hematopoietic stem cell line or all three cell lines. These etiologies involve the following:
· A decrease in or damage to the hematopoietic stem cells and their microenvironment, resulting in hypoplastic or aplastic bone marrow
· Maturation defects, such as in vitamin B-12 or folate deficiency
· Differentiation defects, such as myelodysplasia
· Damage to hematopoietic stem cells can be congenital or acquired. Mechanisms include the following:
· An acquired stem cell injury from viruses, toxins, or chemicals (e.g., chloramphenicol, insecticides) that leads to a quantitative or qualitative abnormality
· Abnormal humoral or cellular control of hematopoiesis
· An abnormal or hostile marrow microenvironment
· Immunologic suppression of hematopoiesis (i.e., mediated by antibodies, T cells, or lymphokines)
BONE MARROW FAILURE
- Mutations in genes causing inherited bone marrow failure syndromes
- The genetic abnormalities in the inherited bone marrow failure syndromes (IBMFS) include:
- Fanconi anemia
- Dyskeratosis congenita (hereditary)
- Shwachman-Diamond syndrome
- Diamond-Blackfan anemia
- Amegakaryocytic thrombocytopenia
- Congenital neutropenia
- Pure red cell aplasia may be a secondary disorder caused by a thymoma. It may also occur transiently from a viral infection, as with parvovirus B19. Pure red cell aplasia may also be permanent as a result of viral hepatitis. It may also occur in lymphoproliferative diseases (e.g., lymphomas, CLL (chronic lymphocytic leukemia)) or collagen vascular diseases (e.g., systemic lupus erythematosus (SLE), refractory anemia), or during pregnancy.
- Amegakaryocytic thrombocytopenic purpura has been reported to occur because of causes similar to those for pure red cell aplasia.
- Single cytopenias: early forms of myelodysplastic syndromes (MDS) can initially present as a single cytopenia or, more often, as a bicytopenia.
- Pancytopenia: a decrease in all three cell lines is the most common manifestation of bone marrow failure. Aplastic or hypoplastic anemia can be idiopathic or secondary. Myelodysplastic anemia can also cause pancytopenia. Myelophthisic anemia may result from marrow destruction because of tumor invasion or granulomas.
- Presentation: Signs & symptoms: Patients with bone marrow failure present with low blood counts. Low platelet counts predispose patients to spontaneous bleeding from the skin and mucous membranes. Neutropenia places the patient at risk for severe infections. Bleeding complications are usually the most alarming symptom, and infections prompt individuals to visit the emergency department. Weakness and fatigue resulting from anemia can develop slowly.
- Lab features:
- Anemia is common, and red cells appear morphologically normal. The reticulocyte count usually is less than 1%, indicating a lack of red cell production. The mean cell volume (MCV) is occasionally elevated, with macrocytosis.
- Platelet counts are lower than normal, with a paucity of platelets in the blood smear. Platelet size is normal, but a low platelet count may increase size heterogeneity.
- Agranulocytosis (i.e., a decrease in all granular white blood cells, including neutrophils, eosinophils, and basophils) and a decrease in monocytes are observed.
- A relative lymphocytosis occurs (i.e., an increased percentage) without an increase in cell count.
- Acquired idiopathic aplastic anemia is usually permanent and life-threatening. Half of the patients die during the first 6 months.
- Bone marrow aspiration & biopsy: A bone marrow aspiration and biopsy should be performed to assess cellularity and the morphology of residual cells. The marrow is replaced with fat cells; stromal cells are replaced with lymphocytes and very few hematopoietic cells. Occasionally, localized pockets of marrow are present (e.g., due to sampling error), which can be misleading. The core biopsy specimen should be at least 1 cm long to evaluate cellularity. Residual erythroid cells may show evidence of dysplasia with nuclear-cytoplasmic maturation dissociation (commonly described, in the absence of a folate or vitamin B-12 deficiency, as megaloblastic features).
- Treatment:
- Supportive: If clinically indicated, initiate a blood transfusion with specific components, such as packed red blood cells for anemia and platelets for thrombocytopenia. Clinical indications for red cell transfusions include symptoms secondary to anemia and bleeding due to thrombocytopenia. Supportive care only temporarily relieves symptoms and does not treat the primary disease.
- Oral aminocaproic acid may easily control bleeding from the nose, gums, or teeth.
- Bone marrow transplantation (BMT): BMT candidates are patients younger than 55 years with severe disease and a matched, related donor. With current BMT regimens, most patients with severe aplastic anemia have a 60 –70% long-term survival rate. Survival rates exceeding 80% have been reported for patients in more favorable subgroups. Using matched, unrelated donors is still less desirable (11 – 20% survival rates).
- Medications:
- Anti-thymocyte globulin (ATG) or anti-lymphocyte globulin (ALG)
- Hematopoietic growth factors, such as granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF), may be useful in patients with neutropenia and infections, without requiring a white blood cell transfusion.
- Immunosuppressants: lymphocyte immune globulin, cyclosporine A, corticosteroids (methylprednisolone, prednisone).
- Androgens: Androgens drive resting hematopoietic stem cells into the cell cycle, making them more responsive to differentiation by hematopoietic growth factors. They also stimulate the endogenous secretion of erythropoietin. Danazol is a non-masculinizing androgen that may be useful. The response rate is limited to approximately 45%, and results may require 6-10 months of therapy.
- Prognosis: The prognosis for bone marrow failure depends on the duration of the marrow dysfunction. Most inherited forms of bone marrow failure, such as Fanconi anemia, are associated with transformation into leukemia several years later. Viral causes, such as parvoviruses, are usually self-limiting.
JAUNDICE/ HYPERBILIRUBINEMIA
· Jaundice (overview): jaundice (also known as icterus) is a yellowish pigmentation of the skin, the conjunctival membranes over the sclerae (whites of the eyes), and other mucous membranes caused by high blood bilirubin levels. This hyperbilirubinemia subsequently causes increased levels of bilirubin in the extracellular fluid. The concentration of bilirubin in blood plasma is normally below 1.2 mg/dl (<25 μmol/L). A level higher than approximately 3 mg/dL (>50 µmol/L) leads to jaundice. Jaundice may be prehepatic, hepatocellular (hepatic), or post-hepatic.
· Pre-hepatic jaundice: it is caused by anything which causes an increased rate of hemolysis, i.e., the breakdown of red blood cells (RBCs). Unconjugated bilirubin comes from the breakdown of the heme pigment found in red blood cells' hemoglobin. The increased breakdown of red blood cells leads to an increase in the amount of unconjugated bilirubin present in the blood, and deposition of this unconjugated bilirubin into various tissues can lead to a jaundiced appearance. Kernicterus is associated with increased unconjugated bilirubin; neonates are especially vulnerable to this due to increased blood-brain barrier permeability. Unconjugated hyperbilirubinemia results from a derailment of the necessary bilirubin conjugation in the hepatocyte (liver cell). This problem may occur before bilirubin has entered the hepatocyte or within the liver cell. Excessive heme metabolism from hemolysis or reabsorption of a large hematoma results in significant increases in bilirubin, which may overwhelm the conjugation process and lead to a state of unconjugated hyperbilirubinemia. Hemolytic anemias usually result in mild bilirubin elevation to about 5 mg/ dL (85.5 μmol/ L), with or without clinical jaundice.
· Causes of prehepatic jaundice: hemolysis. Hemolytic anemias result from abnormal red blood cell survival times. These anemias may occur because of membrane abnormalities (e.g., hereditary spherocytosis); enzyme abnormalities (e.g., glucose-6-phosphate dehydrogenase (G6PD) deficiency); autoimmune disorders, drugs, and defects in hemoglobin structure such as sickle cell disease and the thalassemia; hemolytic uremic syndrome (HUS); malaria, etc. Other causes include defects in bilirubin metabolism, such as Gilbert’s syndrome and Crigler – Najjar syndrome. On the FBC/ CBC (full/ complete blood count), hemolysis is indicated by the presence of fractured red blood cells (schistocytes) and increased reticulocytes on the smear.
· Jaundice – table of diagnostic tests:
· a) Prehepatic jaundice: total bilirubin: normal or increased; conjugated bilirubin: normal; unconjugated bilirubin: normal or increased; urobilinogen: normal or increased; urine color: normal; stool color: normal; ALP (alkaline phosphatase) levels: normal; ALT & AST levels: normal; conjugated bilirubin in urine: not present; splenomegaly: present.
· Features of hemolysis: in hemolysis unconjugated bilirubin is increased (while conjugated bilirubin is normal); urinary urobilinogen is increased; urine color is normal; stool color is normal; ALP (alkaline phosphatase) levels are normal; AST (SGOT) & ALT (SGPT) levels are normal; conjugated bilirubin in urine is not present; splenomegaly may be present. On the FBC/ CBC (full/ complete blood count), hemolytic anemia is indicated by decreased hemoglobin and/or hematocrit (a sign of anemia) and by increased reticulocytes present on the peripheral blood smear (polychromatophilia). On hemolysis, there are also elevated levels of LDH lactate dehydrogenase and decreased levels of plasma haptoglobin. The Direct antiglobulin test (DAT; also called direct Coombs test) is positive in autoimmune hemolytic anemia. Other features of hemolytic anemia include increased free serum hemoglobin, hemoglobin in the urine (hemoglobinuria), and hemosiderin present in the urine (hemosiderinuria).
· In autoimmune hemolytic anemia and hereditary spherocytosis, there is spherocytosis (presence of spherocytes) in the peripheral blood smear.
· Fractured red blood cells (schistocytes) in the blood smear are indicative of microangiopathic hemolytic anemia (MAHA) because of intravascular hemolysis from causes including disseminated intravascular coagulopathy (DIC), thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS). Moreover, methemalbuminemia (the presence of methemalbumin in the circulating blood) is indicative of intravascular hemolysis.
· b) Hepatic jaundice: total bilirubin: increased; conjugated bilirubin: increased; unconjugated bilirubin: increased; urobilinogen: decreased; urine color: dark; stool color: normal or pale; ALP (alkaline phosphatase) levels: increased; ALT & AST levels: increased; conjugated bilirubin in urine: present; splenomegaly: present.
· c) Post-hepatic jaundice: total bilirubin: elevated; conjugated bilirubin: increased; unconjugated bilirubin: normal; urobilinogen: decreased or negative; urine color: dark (conjugated bilirubin); stool color: pale; ALP (alkaline phosphatase) levels: increased; ALT & AST levels: increased; conjugated bilirubin in urine: present; splenomegaly: absent.
· Neonatal jaundice: is a yellowing of the skin and other tissues of a newborn infant. A bilirubin level > 85 μmol/l (5 mg/dL) leads to a jaundiced appearance in neonates [whereas in adults, a level of 34 μmol/l (2 mg/dL) is needed for this to occur]. It is often seen in infants around the second day after birth, lasting until day 8 in normal births or around day 14 in premature births.
· Physiological jaundice: Physiologic jaundice is clinically obvious in 50% of neonates during the first 5 days of life. In physiologic jaundice, the peak total serum bilirubin level is 5 – 6 mg/dL (86 – 103 µmol/L), occurs at 48 – 120 hours (2nd – 5th day) of age and does not exceed 17 – 18 mg/dL (291 – 308 µmol/L). Characteristics: bilirubin is < 20 mg/dl; resolves early. Physiological jaundice is a common condition in which most infants develop visible jaundice due to the elevation of unconjugated bilirubin concentration during their first week. This pattern of hyperbilirubinemia has been classified into two functionally distinct periods:
· a) Phase one:
· i) Term infants: jaundice lasts for about 10 days with a rapid rise of serum bilirubin up to 204 μmol/l (12 mg/dL).
· ii) Preterm infants: jaundice lasts for about two weeks, with a rapid rise of serum bilirubin up to 255 μmol/l (15 mg/dL).
· b) Phase two: bilirubin levels decline to about 34 μmol/l (2 mg/dL) for two weeks, eventually mimicking adult values.
· a) Preterm infants: phase two can last more than one month.
· b) Exclusively breastfed infants: phase two can last more than one month.
· Pathological jaundice: In maternal serum jaundice (Lucey-Driscoll syndrome), jaundice occurs during the first 4 days of life. Characteristics: bilirubin is > 20 mg/dl; kernicterus is common; and resolves late.
· Typical causes for neonatal jaundice include normal physiologic jaundice, jaundice due to formula supplementation, and hemolytic disorders that include hereditary spherocytosis, G6PD deficiency, pyruvate kinase deficiency, ABO or Rh blood type autoantibodies, or infantile pyknocytosis. Serum bilirubin normally drops to a low level without any intervention required. When bilirubin rises, a brain-damaging condition known as kernicterus can occur, leading to significant disability. A Bili light is used for early treatment, which often consists of exposing the baby to intensive phototherapy.
· Note: Two types of jaundice may occur in newborns who are breastfed. Both types are most often harmless.
· a) Breastfeeding jaundice is seen in breast-fed babies during the first week of life. It is more likely to occur when babies do not nurse well, or the mother's milk is slow to come in.
· b) Breast milk jaundice may appear in some healthy, breastfed babies after day 7 of life. It is likely to peak during weeks 2 and 3 but may last for a month or more at low levels. The problem may be due to how substances in breast milk affect the breakdown of bilirubin in the liver. Breast milk jaundice is different than breastfeeding jaundice. In breast milk jaundice, the bilirubin may increase to levels as high as 20 mg/dL, necessitating the need for phototherapy and the discontinuation of breastfeeding.
{kind=link}
{kind=link}
· Hyperbilirubinemia in the term newborn: neonatal hyperbilirubinemia is defined as a total serum bilirubin level above 5 mg per dL (86 μmol per L). Although up to 60% of term newborns have clinical jaundice in the first week of life, few have a significant underlying disease. However, hyperbilirubinemia in the newborn period can be associated with severe illnesses such as hemolytic disease, metabolic and endocrine disorders, anatomic abnormalities of the liver, and infections.
· Jaundice typically results from the deposition of unconjugated bilirubin pigment in the skin and mucous membranes. Depending on the underlying etiology, this condition may present throughout the neonatal period.
· Unconjugated hyperbilirubinemia is the most common form of jaundice encountered by family physicians.
· Causes of neonatal hyperbilirubinemia can be classified into three groups based on the mechanism of accumulation: bilirubin overproduction, decreased bilirubin conjugation, and impaired bilirubin excretion.
· Ι) Increased bilirubin load:
· a) Hemolytic causes: Characteristics: increased unconjugated bilirubin level, >6% reticulocytes, hemoglobin concentration of <13 g/dL (130 g/ L).
· i) Coombs' test positive: Rh factor incompatibility, ABO incompatibility, minor antigens incompatibility.
· ii) Coombs' test negative: red blood cell (RBC) membrane defects (spherocytosis, elliptocytosis), RBC enzyme defects (G6PD deficiency, pyruvate kinase deficiency), medications (e.g., sulfisoxazole acetyl with erythromycin ethyl succinate, streptomycin, vitamin K), abnormal RBCs (hemoglobinopathies), sepsis.
· b) Non-hemolytic causes Characteristics: increased unconjugated bilirubin level, normal percentage of reticulocytes.
· i) Extravascular sources: cephalohematoma, bruising, central nervous system (CNS) hemorrhage, swallowed blood.
· ii) Polycythemia: fetal-maternal transfusion, delayed cord clamping, twin-twin transfusion.
· iii) Exaggerated enterohepatic circulation: cystic fibrosis, ileal atresia, pyloric stenosis, Hirschsprung's disease, breast milk jaundice.
· II) Decreased bilirubin conjugation: Characteristics: increased unconjugated bilirubin level, normal percentage of reticulocytes. Causes: physiologic jaundice; Crigler-Najjar syndrome types 1 and 2; Gilbert syndrome; hypothyroidism; breast milk jaundice.
· III) Impaired bilirubin excretion: Characteristics: increased unconjugated and conjugated bilirubin level, negative Coombs' test, conjugated bilirubin level of >2 mg/dL (34 μmol/L) or >20% of total serum bilirubin level, conjugated bilirubin in urine.
· Causes:
· a) Biliary obstruction: biliary atresia, choledochal cyst, primary sclerosing cholangitis, gallstones, neoplasm, Dubin-Johnson syndrome, Rotor's syndrome.
· b) Infection: sepsis, urinary tract infection, syphilis, toxoplasmosis, tuberculosis, hepatitis, rubella, herpes.
· c) Metabolic disorder: alpha 1 antitrypsin deficiency, cystic fibrosis, galactosemia, glycogen storage disease, Gaucher's disease, hypothyroidism, Wilson's disease, Niemann-Pick disease.
· d) Chromosomal abnormality: Turner's syndrome, trisomy 18 and 21 syndromes.
· e) Medications: aspirin, acetaminophen, sulfa, alcohol, rifampin, erythromycin, corticosteroids, tetracycline, etc.
· Unconjugated hyperbilirubinemia – causes:
· I) Hemolysis. Hemolytic disorders are either inherited or acquired. Inherited disorders, such as spherocytosis or sickle cell anemia, may be suggested by other laboratory abnormalities, including peripheral blood smear, elevated levels of lactate dehydrogenase (LDH), and decreased levels of haptoglobin. In these settings, serum bilirubin levels rarely exceed 5 mg/dL.
· II) Ineffective erythropoiesis (early labeled bilirubin (ELB) production): onset of asymptomatic jaundice. This is characterized by a marked increase in fecal urobilinogen excretion and a normal or near-normal red blood cell lifespan. A marked increase in ELB formation has been documented in diseases associated with ineffective erythropoiesis, including iron deficiency anemia, pernicious anemia, thalassemia, erythropoietic porphyria, lead poisoning, and physiologic neonatal jaundice.
· III) Crigler-Najjar syndrome type 1: Crigler-Najjar type 1 is an exceptionally rare condition caused by the absence of bilirubin UDP glucuronyltransferase (UGT-1) activity. This syndrome usually develops during the neonatal period and leads to infant death. Apart from jaundice, the affected infant usually appears healthy at birth. Jaundice develops in the first few days of life and rapidly progresses by the second week; patients may present with evidence of kernicterus, the clinical manifestations of which are hypotonia, deafness, oculomotor palsy, lethargy, and, ultimately, death. Therefore, exchange transfusion is warranted despite phototherapy. Apart from jaundice, physical findings are usually normal in Crigler Najjar syndrome type 1, with no signs of hemolysis or liver disease. Except for the presence of high serum unconjugated bilirubin levels, the results of liver tests in Crigler-Najjar syndrome type 1 are normal. Serum bilirubin levels range from 20 – 50 mg/dL. Conjugated bilirubin is absent from serum, and bilirubin is not present in urine. A family history that includes consanguinity, relatives with severe jaundice without hemolysis, or relatives with evidence of liver disease and a history of exchange transfusion further supports the diagnosis. Because of its autosomal recessive transmission, consanguinity is a risk factor for Crigler-Najjar syndrome type 1. Definitive diagnosis of Crigler-Najjar syndrome requires high-performance liquid chromatography (HPLC) of bile or a tissue enzyme assay of a liver biopsy sample.
· IV) Crigler-Najjar syndrome type 2: is more common than type 1 and less severe. The bilirubin UDP glucuronyltransferase (UGT-1) activity is usually markedly reduced; however, phenobarbital increases this activity and reduces jaundice. Patients with this disorder have a normal life expectancy. Usually, no clinical symptoms are reported with this disease. However, bilirubin encephalopathy has been reported. Crigler-Najjar syndrome type 2 patients appear healthy at birth, with no signs of liver disease. Patients may present with evidence of kernicterus, the clinical manifestations of which are hypotonia, deafness, oculomotor palsy, lethargy, and death. Crigler-Najjar syndrome type 2 results in lower bilirubin concentrations than does type I, with levels ranging from 7 – 20 mg/dL.
· V) Gilbert syndrome: the most common of the 3 inherited conditions, is found in 3% to 7% of the American population. The conjugation process is impaired because of reduced bilirubin UDP glucuronosyltransferase activity. This syndrome is benign and rarely produces clinical jaundice. Serum bilirubin levels may rise 2- or 3-fold with fasting or dehydration but are almost always less than 6 mg/dL. It may manifest only as jaundice on clinical examination; at least 30% of patients with Gilbert syndrome are asymptomatic, although nonspecific symptoms, such as abdominal cramps, fatigue, and malaise, are common. Abdominal symptoms may be multifactorial, with underlying anxiety probably playing an important role. Although it is true that not all patients with Gilbert syndrome and abdominal symptoms are anxious, they nonetheless appear to have organic-type discomfort that is hard to characterize and frequently eludes diagnosis. Gilbert syndrome can be diagnosed by a thorough history and physical examination and confirmed by standard blood tests. No relationship exists between abdominal symptoms and plasma bilirubin levels. Apart from mild jaundice, physical examination findings in people with Gilbert syndrome are normal. Lab results include unconjugated hyperbilirubinemia noted on several occasions; normal results from a full/complete blood count (FBC/CBC), reticulocyte count, and blood smear; normal liver function test (LFT) results; absence of other disease processes. Diagnosis is made in patients who have no past history of liver disease and manifest only jaundice on clinical examination. Specialized tests that have occasionally been used to confirm a diagnosis of Gilbert syndrome include fasting test, nicotinic acid test, phenobarbital test, radiolabeled chromium test, thin-layer chromatography, drug clearance test, polymerase chain reaction (PCR) assay, percutaneous liver biopsy (very rarely performed). Note: Rarely, certain drugs may decrease bilirubin uptake. A prime example is rifampin, which may exacerbate unconjugated hyperbilirubinemia in those with Gilbert syndrome.
· VI) Physiologic neonatal jaundice: clinically obvious in 50% of neonates during the first 5 days of life. In physiologic jaundice, the peak total serum bilirubin level is 5 – 6 mg/dL (86 – 103 µmol/L), occurs at 48 – 120 hours of age and does not exceed 17 – 18 mg/dL (291 – 308 µmol/L).
· VII) Non-physiologic neonatal jaundice: In maternal serum jaundice (Lucey-Driscoll syndrome), jaundice occurs during the first 4 days of life.
· VIII) Breast milk jaundice: bilirubin may increase to levels as high as 20 mg/dL, necessitating the need for phototherapy and the discontinuation of breastfeeding.
· Note: physiologic VS non-physiologic neonatal jaundice: While no specific test exists for physiologic jaundice, other causes of jaundice should be considered in infants with one or more of the following: jaundice occurring within 24 hours of birth; serum concentrations of unconjugated bilirubin of 11 – 12 mg/dL in infants who are formula-fed or 14 – 15 mg/dL in infants who are breastfed; increased levels of conjugated bilirubin (>2 mg/dL); jaundice persisting for more than 2 weeks.
· IX) Hematoma resorption.
· X) Other causes: Other conditions to be considered include acute and chronic liver disease; primary hyperbilirubinemia from ineffective erythropoiesis; infections; cardiac disease (such as congestive heart failure or prosthetic heart valves); rhabdomyolysis; high-altitude living; medications (such as probenecid, rifampicin, or other antibiotics); thyrotoxicosis. Other differentials: dyserythropoiesis, portosystemic shunts, ethinyl estradiol.
· Differential diagnosis: iron-deficiency anemia; pediatric lead toxicity; pernicious anemia.
· Jaundice – diagnostic approach (algorithm):
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Hemolytic anemia:
HEMOLYSIS / HEMOLYSIS RELATED DISORDERS
· Hemolysis/ hemolytic anemia (overview): hemolytic anemia occurs when the bone marrow is unable to replace the red blood cells that are being destroyed. Immune hemolytic anemia occurs when the immune system mistakenly sees the person’s own red blood cells as foreign substances. Antibodies then develop against the red blood cells (RBCs). These antibodies attack the red blood cells and cause them to break down too early.
· Causes: Red blood cells may be destroyed due to:
· a) Genetic defects within the red cells, such as sickle cell anemia, thalassemia, and G6PD deficiency.
· b) Exposure to certain chemicals, drugs, and toxins.
· c) Infections.
· d) Blood clots in small blood vessels.
· e) Transfusion of blood from a donor with a blood type that does not match with the recipients.
· Hemolysis – Workup: full/ complete blood count (FBC/ CBC) can help diagnose anemia and offer hints to the problem's type and cause. Essential parts of the CBC include red blood cell count (RBC), hemoglobin, hematocrit (HCT), and platelet (Plt) count. The following tests can identify the type of hemolytic anemia: absolute reticulocyte count; direct antiglobulin test (DAT; also known as direct Coombs test); indirect Coombs test; Donath – Landsteiner test (a test for antibodies related to paroxysmal cold hemoglobinuria); cold agglutinins (a test for cold agglutinin disease); free hemoglobin in the serum or urine; hemosiderin in the urine; serum protein electrophoresis; pyruvate kinase activity (to check for pyruvate kinase deficiency); osmotic fragility test (to exclude hereditary spherocytosis), serum haptoglobin levels; serum LDH, urine, and fecal urobilinogen, and G6PD levels.
· Features of hemolysis: in hemolysis unconjugated bilirubin is increased (while conjugated bilirubin is normal); urinary urobilinogen is increased; urine color is normal; stool color is normal; ALP (alkaline phosphatase) levels are normal; AST (SGOT) & ALT (SGPT) levels are normal; conjugated bilirubin in urine is not present; splenomegaly may be present.
· On the FBC/ CBC (full/ complete blood count), hemolytic anemia is indicated by decreased hemoglobin and/or hematocrit (a sign of anemia) and is also shown by the presence of increased reticulocytes that are also present on the peripheral blood smear.
· On hemolysis, there are also elevated levels of LDH lactate dehydrogenase and decreased levels of plasma haptoglobin.
· Direct antiglobulin test (DAT; also called direct Coombs test) is positive in autoimmune hemolytic anemia. Other features of hemolytic anemia include increased free serum hemoglobin, hemoglobin in the urine (hemoglobinuria), and hemosiderin present in the urine (hemosiderinuria) in intravascular hemolysis.
· In autoimmune hemolytic anemia and hereditary spherocytosis, there is spherocytosis (presence of spherocytes) in the peripheral blood smear.
· Fractured red blood cells (schistocytes) in the blood smear are indicative of microangiopathic hemolytic anemia (MAHA) or other causes for intravascular hemolysis, including disseminated intravascular coagulopathy (DIC)).
· Moreover, methemalbuminemia (the presence of methemalbumin in the circulating blood) is indicative of intravascular hemolysis.
· Note: methemalbumin is an albumin complex consisting of albumin and heme. This complex gives a brown color to plasma and occurs in hemolytic and hemorrhagic disorders. The Schumm test uses spectroscopy to determine significant levels of methemalbumin in the blood. It is used to differentiate intravascular hemolysis from extravascular hemolysis, as in hemolytic anemias. A positive result is indicative of intravascular hemolysis.
· Hemolysis - characteristics: raised reticulocyte count, polychromatic and nucleated RBCs on a blood smear (e.g., on thalassemia major), erythroid hyperplasia in bone marrow, elevated indirect (non-conjugated) serum bilirubin, raised urobilinogen in urine, increased serum LDH. Note: rule out acute blood loss!
· Hemolysis – investigation: check for splenomegaly; blood smear, Hb (hemoglobin) electrophoresis, family studies, osmotic fragility test (e.g., hereditary spherocytosis or elliptocytosis), sucrose lysis test (paroxysmal nocturnal hemoglobinuria), G6PD, malaria, direct Coomb’s (DAT) test; also check for microangiopathic hemolysis.
· Intravascular hemolysis: Causes: incompatible blood transfusion, PNH (paroxysmal nocturnal hemoglobinuria), PCH (paroxysmal cold hemoglobinuria), and blackwater fever (hemolysis in malaria). Fractured red blood cells (schistocytes) in the blood smear are indicative of microangiopathic hemolytic anemia (MAHA) because of intravascular hemolysis from causes including disseminated intravascular coagulopathy (DIC), thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS). There is NO splenomegaly. Moreover, methemalbuminemia (the presence of methemalbumin in the circulating blood) is indicative of intravascular hemolysis.
· Increased: reticulocyte count, indirect (non-conjugated) bilirubin, plasma hemoglobin (markedly increased), serum LDH. Also, hemoglobin in the urine, hemosiderin in urine, methaemalbumin positive (Schumm’s test), and decreased serum haptoglobin.
· Extravascular hemolysis: macrophages of the spleen, liver, bone marrow, etc. Causes: hemoglobinopathies, hereditary hemolytic anemias, autoimmune hemolytic anemia. Splenomegaly may occur. Increased reticulocyte count, indirect (non – conjugated) serum bilirubin, serum LDH, plasma hemoglobin (mild to moderate heightened). Also, absent hemoglobin & hemosiderin in urine, negative for methaemalbumin; and decreased haptoglobin.
· Note: Serum hemoglobin= free-floating Hb (Hbg, hemoglobin) in blood serum.
· HELLP syndrome (on the pregnant): Hemolysis, Elevated Liver function tests (liver enzymes), Low Platelets. It is a variant or complication of pre-eclampsia.
· Hemolytic anemia: low Hg and Hct, increased reticulocyte count, increased indirect (non – conjugated) bilirubin and serum LDH, decreased serum haptoglobin, blood smear (polychromasia, nucleated RBCs).
· Autoimmune hemolytic anemia: Abs (antibodies) against the person’s own RBCs.
· a) Warm antibody: i) Primary – idiopathic (50%). ii) Secondary: autoimmune disorders (SLE (systemic lupus erythematosus), RA (rheumatoid arthritis), UC (ulcerative colitis), scleroderma (systemic sclerosis), etc.), lymphoproliferative disorders (e.g., lymphoma and CLL (chronic lymphocytic leukemia). Less common causes include neoplasms (other than lymphoids) and infections.
· b) Cold antibody: cold agglutinin disease (see below), paroxysmal cold hemoglobinuria (see below).
· c) Drug-related (e.g., antibiotics (penicillin, cephalosporins, levofloxacin, nitrofurantoin), NSAIDs, phenazopyridine (used as an analgesic for the urinary tract), quinidine (antiarrhythmic Ia class), dapsone, methyldopa (for hypertension), levodopa (for Parkinson’s), etc.).
· Clinical features: Anemia, jaundice, splenomegaly.
· Lab features Direct Coombs (DAT; direct antiglobulin test) test positive. Elevated serum indirect (unconjugated) bilirubin, increased urine urobilinogen, reduced plasma haptoglobin, and increased serum LDH, and reticulocytosis. Also, methemalbuminemia, hemoglobinuria, and hemosiderinurea in cold agglutinin disease and paroxysmal cold hemoglobinuria where hemolysis is intravascular.
· Blood smear: e.g., spherocytes, polychromatic cells, normoblasts.
· Alloimmune hemolytic anemia: e.g., hemolytic disease of the newborn, hemolytic transfusion reaction (incompatibility).
· Cold agglutinin disease (CAD): autoimmune disease, elevated levels of circulating Abs (antibodies), usually IgM, against RBCs. It is a form of autoimmune hemolytic anemia.
· It is caused by cold-reacting autoantibodies that bind to the erythrocyte membrane, leading to premature erythrocyte destruction (hemolysis) (autoimmune hemolytic anemia).
· Antibodies bind RBCs only at low body temperatures, typically 28 – 31 Celsius degrees.
· Autoagglutination on the blood smear.
· Cold agglutinins are antibodies that are usually specific for I antigen, an RBC surface carbohydrate macromolecule. Cold agglutinins bind to the erythrocyte surface antigen at a temperature optimum of 0 – 4 °C, which causes agglutination of erythrocytes and, thereby, impaired microcirculation ranging from moderate acrocyanosis to severe Raynaud’s phenomenon. The antigen-antibody complex also induces the classical complement pathway, resulting in extravascular hemolysis. The physiologic cooling of blood in peripheral vessels is usually sufficient to cause hemolysis and circulatory symptoms in patients with a high thermal amplitude of cold agglutinins. The clinical effects of cold agglutinins depend on the antibody's titer and thermal amplitude. Cold agglutinins are rare, and only low titers can be found in the serum of healthy individuals. Cold agglutinins may be monoclonal or polyclonal. Monoclonal antibodies are usually detected in patients with idiopathic forms of cold agglutinin disease or lymphoproliferative disorders. Polyclonal antibodies typically appear after infection, most often with Mycoplasma pneumonia, EBV, or CMV. Cold agglutinins can cause hemolysis in patients undergoing cardiac surgery during a hypothermic cardiopulmonary bypass. In cold agglutinins, Hematocrit may appear to decrease, and MCV elevated (Reference: http://www.biochemia-medica.com/2014/24/391 ).
· Often, the first indication of the existence of cold agglutinins is the failure to obtain a meaningful RBC count and indices. The hemoglobin and hematocrit results do not match. The RBC count will be decreased due to doublet erythrocytes being counted as a single cell, thus resulting in a falsely high MCV. Hematocrit will also be lowered, as the volume of doublets is slightly less than 2 cells. The MCHC and MCH values will be increased due to decreased hematocrit and RBC count. Invalid red blood cell indices can also be due to lipemia, hemolysis, icterus, or hereditary spherocytosis (Reference: http://labmed.oxfordjournals.org/content/labmed/33/6/455.full.pdf ).
· The combination of low hematocrit, normal hemoglobin, and high MCV and MCHC is characteristic of cold agglutinins. Practically, hematocrit is not equal to the triple of hemoglobin but much lower; often, its value is close to that of the hemoglobin.
· Causes of cold agglutinin disease: primary (idiopathic) or secondary infections (e.g. Mycoplasma pneumonia, viral pneumonia and other respiratory infections, infectious mononucleosis (EBV or CMV), other viral infections, (mumps, varicella, rubella, adenovirus, HIV, influenza, hepatitis C), bacterial infections (Legionnaire disease, syphilis, listeriosis, Escherichia coli), parasitic infections (malaria, trypanosomiasis), lymphoproliferative disorders (e.g. lymphoma, chronic lymphocytic leukemia, Waldenstrom's macroglobulinemia, multiple myeloma), non – hematological neoplasms, CANOMAD syndrome (chronic ataxic neuropathy ophthalmoplegia M-protein agglutination disialosyl antibodies), chromosomal anomalies (trisomy 3 and trisomy 12), patients after living-donor liver transplantation treated with tacrolimus and after bone marrow transplantation with cyclosporine treatments, patients with sclerodermic features, patients with hyperreactive malarial splenomegaly (HMS) (an immunopathologic complication of recurrent malarial infection), Diphtheria-pertussis-tetanus (DPT) vaccination (IgM autoantibody with a high thermal range), Equestrian perniosis (a rare cause), patient subjected to hypothermia for cardiopulmonary bypass surgery (rare).
· Symptoms: A common complaint among patients with cold agglutinin disease is sore fingers and toes with purplish discoloration associated with cold exposure. In chronic cold agglutinin disease, the patient is more symptomatic during the colder months. Cold agglutinin–mediated acrocyanosis differs from the Raynaud phenomenon. In Raynaud phenomena caused by vasospasm, a triphasic color change occurs, from white to blue to red, based on vasculature response. No evidence of such a response exists in cold agglutinin disease.
· Other symptoms of cold agglutinin disease include respiratory symptoms (e.g., in patients with Mycoplasma pneumoniae infection), hemoglobinuria (results from hemolysis; this may be reported following prolonged exposure to cold; hemoglobinuria is more commonly seen in paroxysmal cold hemoglobinuria), chronic fatigue (due to anemia).
· CBC (complete blood count): If the cold agglutinin is operative at room temperature, then a falsely high mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), and mean corpuscular hemoglobin concentration (MCHC) with a low RBC count is obtained due to agglutination of RBCs in the cold, automated counter.
· Agglutination may also be seen in anticoagulated blood at room temperature. This agglutination worsens with storage and cooling of blood to 4°C and disappears rapidly upon warming the blood to 37°C, unlike with rouleaux formation. Repeating the complete blood count (CBC) after warming the blood to 37°C avoids this problem. Thus, the clinical laboratory is frequently the first to report the presence of a cold agglutinin. Agglutination in the cold may also interfere with typing and cross-matching of blood.
· In patients with chronic cold agglutinin disease, mild to moderate, stable anemia is present; occasionally, the anemia is severe.
· Peripheral blood smears may reveal clumps of RBCs. Leukocytosis may be evident during hemolytic episodes. Spherocytes may be present, although less prominently than warm autoantibody–induced hemolytic anemias.
· The results of the reticulocyte count are usually increased in patients with cold agglutinin disease, with polychromasia in the peripheral blood smear.
· The MCV is elevated because of reticulocytosis and agglutination of the RBCs. However, reticulocytosis may be inadequate for the degree of anemia in the patient. This may be due to decreased erythropoiesis caused by an underlying infection.
· Signs of hemolysis: LDH and total and direct bilirubin values are elevated, depending on the extent of hemolysis. Urinalysis can reveal hemoglobinuria, hemosiderinuria, and elevated urobilinogen. The haptoglobin level may be reduced when hemolysis is active.
· The direct antiglobulin test (DAT; direct Coomb’s) should be performed with samples at 35-37°C, using polyspecific and monospecific Coombs sera, including monospecific anti-C3 and IgG antisera. Coombs testing is positive for anti-C3 and classically negative anti-IgG.
· Cryoglobulin levels should be tested only if vascular purpura or other atypical findings, such as elevated levels of IgM and/or hepatitis virus antibodies, are found. Again, proper handling of the sample, by keeping it warm until the test is run, is essential to avoid premature loss of the cryoglobulin.
· On testing the cold agglutinin titer, a titer greater than 1:64 is considered abnormal when blood is tested at 4°C. The cold agglutinin titer should also be obtained at 30°C and 37°Celcious when needed. Testing at temperatures higher than 4°C is extremely valuable, particularly if the patient is to undergo hypothermia for surgery. Cold agglutinin disease is usually associated with very high cold agglutinin titers of greater than 1:10,000 at 4°C, with a thermal amplitude of up to 30-32°C. The addition of bovine serum albumin (BSA) while testing for the cold agglutinin titer and thermal amplitude results in a better correlation with clinical hemolytic anemia than does obtained data in the absence of BSA using saline-suspended cells.
· Differential diagnosis with Cryoglobulinemia: cryoglobulinemia has almost none of the features of cold agglutinin disease, except for a history of Raynaud syndrome and an elevated IgM level in some cases (but without hemolysis).
· Treatment: In most cases, cold agglutinin disease may be managed successfully using protective measures (clothing) alone. Therapy is directed at serious symptoms and the underlying disorder if any is found. Chemotherapeutic agents should be used under appropriate circumstances, such as for an associated malignancy. However, the use of chemotherapeutic or immunosuppressive agents in the routine management of idiopathic cold agglutinin disease because of its basic benign nature.
· The anti-CD20 monoclonal antibody rituximab depletes B-lymphocytes, thereby interfering with the production of a cold agglutinin. In studies, patients with cold agglutinin disease responded promptly to the drug. Glucocorticoids are generally not useful in IgM-induced cold agglutinin disease but may occasionally work if an underlying warm antibody-induced hemolytic anemia exists; if a high thermal amplitude, low titer cold agglutinin is present or (rarely) if a cold-reactive IgG antibody is produced.
· Plasmapheresis effectively, albeit temporarily, removes IgM antibody from plasma, reducing its concentration. This procedure is valuable for emergencies and allows time for drugs to have an effect.
· Direct antiglobulin test (DAT; also called direct Coombs test): it detects Abs (antibodies) bound in vivo to RBCs. In the test, the RBCs of the patient are mixed with an antiglobulin reagent (anti–IgG Abs). Agglutination of RBCs indicates a positive test.
· Indirect antiglobulin test (also called indirect Coombs test): it detects Abs (antibodies) bound in vitro to RBCs. In the test, the patient's serum (that contains IgG Abs) is mixed with normal group O or the donor’s RBCs. The RBCs are then coated with IgG Abs and react with Coombs, antiserum (Anti – IgG Abs). Agglutination of RBCs indicates a positive test.
· Rhesus hemolytic disease of the newborn (Rh HDN): Mother Rh (D) negative, father Rh (D) positive, baby Rh (D) positive. Blood smear of the newborn: many polychromatic cells & erythroblasts. Elevated indirect (non - conjugated) serum bilirubin. Positive Coombs (antiglobulin) test. Anti – D Ab (antibody). Kleihauer-Betke test: detects fetomaternal hemorrhage by detecting fetal cells in maternal circulation (in the test, fetal RBCs are dark, while maternal are pale and empty).
· Hereditary spherocytosis: presentation in children with anemia, jaundice & splenomegaly.
· Gallstones are common.
· CBC/ blood smear: Blood smear shows spherocytes. Also, there is reticulocytosis.
· Diagnosis: osmotic fragility test, flow cytometry of labeled intact RBCs, and acidified glycerol lysis test.
· G6PD deficiency: asymptomatic in steady-state; hemolytic anemia on exposure to oxidant stress, hemolysis (hemoglobinuria, reticulocytosis, increased LDH, and indirect (non – conjugated) bilirubin, decreased haptoglobin).
· Negative direct Coombs (DAT) test.
· Blood smear: bite cells (also called degmacytes) (Heinz bodies on RBCs that are removed by splenic macrophages; methyl violet staining), blister cells (when the red cell membrane around the bite repairs, a blister-like structure forms, hence the term blister cell), half-ghost cells.
· Diagnosis: Beutier fluorescent spot test (2 – 3 weeks after the hemolytic episode) and methemoglobin reduction test.
· ITP (idiopathic thrombocytopenic purpura):
· Clinical features: Abrupt (children) or gradual (adults) onset, purpura, bruising, bleeding diathesis, e.g., menorrhagia, epistaxis, gingival bleeding. Recent viral illness or live virus vaccine (children). Decreased platelets (giant platelets in congenital thrombocytopenia).
· Pathogenesis: IgG Abs (antibodies) on Platelet surface.
· Treatment: glucocorticoids, IV Ig (IV immunoglobulin), platelet transfusion in severe hemorrhage.
· Differential diagnosis: drugs (heparin, alcohol, quinine, quinidine), infections (e.g., HIV).
· Thrombotic thrombocytopenic purpura (TTP; also known as Moschcowitz syndrome): decreased platelets, MAHA (microangiopathic hemolytic anemia), fluctuating neurological signs, renal impairment, fever.
· MAHA (microangiopathic hemolytic anemia): increased LDH, increased indirect (unconjugated) bilirubin, and schistocytes on the blood smear.
· Fluctuating neurological signs: e.g., hallucinations, altered mental status, headache, stroke, bizarre behavior, seizures, hemiplegia, aphasia, visual problems, paresthesia, TIA (transient ischemic attack), transient sensorimotor deficits, etc.
· Treatment: plasmapheresis (exchange transfusion), corticosteroids, rituximab (if resistant).
· Hemolytic uremic syndrome (HUS): hemolytic anemia, AKI (acute kidney injury), thrombocytopenia.
· Predominantly, but not exclusively, on children.
· Most cases are preceded by infectious diarrhea (which may be bloody), e.g., E. coli (especially the Ο157: Η7 serotype), Campylobacter, Shigella, or viruses.
· Mortality 5 – 10%.
· A minority develops chronic kidney disease.
· Signs/ symptoms: oliguria, hematuria, kidney failure, low platelets, hypertension.
· Also, microangiopathic hemolytic anemia (MHA): increased LDH, increased indirect (unconjugated) bilirubin, and schistocytes on the blood smear.
· Neurological signs may also occur in some cases.
· Treatment is supportive, e.g., dialysis on renal failure.
· Note: HUS & TTP belong to the same spectrum that causes MHA (microangiopathic hemolytic anemia).
· Microangiopathic hemolytic anemia (MAHA): A microangiopathic subgroup of hemolytic anemia. There is intravascular fragmentation and lysis of RBCs due to alteration in small blood vessels.
· Common causes of ΜΑHA: severe infections, DIC (disseminated intravascular coagulation), disseminated malignancy, TTP (thrombotic thrombocytopenic purpura), HUS (hemolytic uremic syndrome), malignant hypertension, pro/eclampsia, SLE (systemic lupus erythematosus), cancer and prosthetic heart valve
· Blood smear: shows schistocytes.
· Mechanical hemolytic anemia: mechanical hemolytic anemia is a form of hemolytic anemia due to mechanically induced damage to RBCs. Red blood cells, while flexible, may, in some circumstances, succumb to physical shear and compression, resulting in hemoglobinuria. The damage is induced through repetitive mechanical motions such as prolonged marching, known as ‘march hemoglobinuria,’ marathon running/ ‘runner’s macrocytosis’ (the impact forces from running can lead to red blood cell hemolysis and accelerate red blood cell production; thus, there may be hemolysis and increased MCV on runners), and bongo drumming (bongo drums are Afro – Cuban drums). Mechanical damage can also be induced through the chronic condition of microangiopathic hemolytic anemia due to prosthetic heart valves.
· Paroxysmal nocturnal hemoglobinuria (PNH): rare; genetically acquired, destruction of RBCs by the component, intravascular hemolytic anemia. Abnormal sensitivity of RBCs to the hemolytic action of the compliment.
· Intravascular hemolysis, anemia, bone marrow failure, thrombosis.
· May occur progression to aplastic anemia and acute leukemia.
· Pathogenesis: normal RBCs are protected from complement activation by the 2 membrane proteins DAF (CD25) and MIRL (CD59) that are anchored to the red cell membrane by GPI anchor. On PNH GPI anchor is not factional due to the genetic mutation in the PIG-A gene, leading to the deficiency (due to non-anchorage) of CD55 & CD59 on the RBC membrane. Thus, complement bounds to the RBC’s membranes, it is not inactivated, and the RBCs are destroyed.
· Characteristic: High incidence of thrombosis, DVT (deep vein thrombosis), hepatic vein (Budd – Chiari syndrome), portal vein, mesenteric vein, cerebral veins).
· Signs & symptoms: red discoloration of urine (destruction of RBCs, the presence of hemoglobin and hemosiderin), especially in the morning (when the urine is more concentrated), symptoms of anemia (palpitations, tiredness, shortness of breath (SOB)).
· Lab tests: Pancytopenia, anemia, hemoglobinemia, hemolysis [increased LDH, increased indirect (non – conjugated) bilirubin, increased reticulocytes, decreased haptoglobin], and anemia. Also, hemoglobinuria, and hemosiderinuria (we have intravascular hemolysis).
· Negative DAT (direct Coombs).
· Diagnosis: Ham’s test, sucrose lysis test (old test). Gold standard: flow cytometry (levels of CD55 & CD59) & FLAER (fluorescein-labeled proaerolysin) test.
· Treatment: steroids? +_ transfusion, warfarin (thrombosis prevention), eculizumab.
· Evans syndrome: an autoimmune disease where the individual’s Abs (antibodies) attack their own RBCs & platelets. Both events may co-occur, or one may follow on from the other. Other antibodies may occur directed against neutrophils and lymphocytes. Its overall pathology resembles a combination of autoimmune hemolytic anemia and idiopathic thrombocytopenic purpura. About 10 – 23% of patients who have autoimmune hemolytic anemia will also have thrombocytopenia and, thus, Evans syndrome.
· Diagnosis: blood tests to confirm not only hemolytic anemia and idiopathic thrombocytopenic purpura. Also, a positive DAT (direct Coombs) and an absence of any known underlying cause.
· Treatment: initial treatment is with corticosteroids or IV (intravenous) immunoglobulin. Relapses are not uncommon, and immunosuppressive medications may be used. Other treatment options include rituximab (in acute and refractory cases) and, in some cases, splenectomy. The only prospect for a permanent cure is the high-risk option of an allogenic hematopoietic stem cell transplantation (SCT).
· DIC (disseminated intravascular coagulation): an acquired disorder that occurs in a wide spectrum of underlying conditions.
· Characteristics:
· i) Widespread systemic activation of coagulation with the formation of microthrombi (small clots) in small blood vessels.
· ii) Bleeding diathesis secondary to depletion of coagulation factors and platelets.
· Mechanism: activation of coagulation factors and platelets; formation of microthrombi in circulation; end-organ damage and depletion of coagulation factors and platelets due to their consumption leading to bleeding diathesis.
· Causes: sepsis/ severe infections, trauma (especially brain or crush injury), obstetric problems (amniotic fluid embolism, abruption placenta, septic abortion, eclampsia, intrauterine retention of dead fetus), malignancy (disseminated solid cancer, acute promyelocytic leukemia), severe hemolytic transfusion reactions, thermal injury (heat stroke, extensive burns), snakebite (e.g., Russell’s viper), severe liver disease, giant hemangioma (Kasabach – Merritt syndrome). Also, AML (acute myeloid leukemia) subtype M3 (APL, acute promyelocytic leukemia) chemotherapy treatment may be complicated when the promyelocytes release the contents of their granules into the peripheral circulation.
· For DIC causes, see: http://emedicine.medscape.com/article/199627-overview#a7
· Clinical features: sudden onset of spontaneous bleeding from multiple sites (petechia, purpura, hematemesis, melena, hematuria, epistaxis, oozing from venipuncture sites); purpura fulminans (patchy areas of hemorrhagic skin necrosis) and presence of an underlying condition associated with DIC.
· Lab features: reduced platelets or falling platelets on repeat testing; increased D – dimmers, increased FDPs (fibrinogen/ fibrin degradation products); prolonged PT & aPTT; reduced Fibrinogen or falling level on repeat testing. (but, as fibrinogen is an acute phase reactant, it may be normal or even increased by 57%).
· The massive fibrin deposition in DIC suggests that fibrinogen levels would be decreased. Thus, the measurement of fibrinogen has been widely advocated as a useful tool for the diagnosis of DIC; however, it is, in fact, not very helpful. Fibrinogen, as a positive acute-phase reactant, is increased in inflammation, and whereas values may decrease as the illness progresses, they are rarely low. One study demonstrated that in up to 57% of DIC patients, the levels of fibrinogen may, in fact, remain within normal limits.
Low fibrinogen may be caused by liver failure.
· Also, low plasma levels of coagulation inhibitors (ATIII (antithrombin III) or protein C).
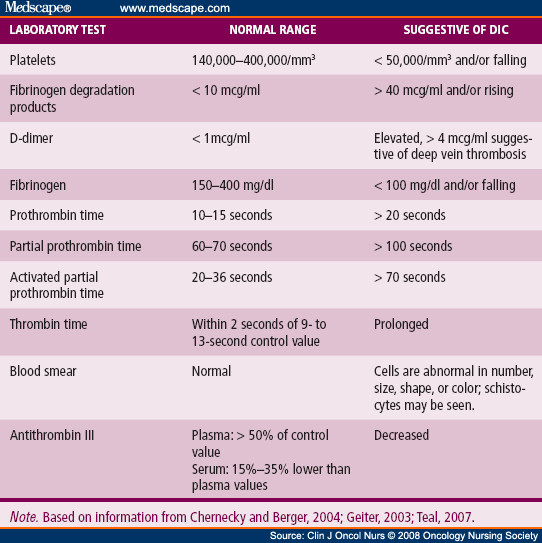{kind=link}
· Blood smear may show schistocytes (fragmented red cells), indicative of microangiopathic hemolytic anemia (MAHA) (intravascular hemolysis).
· The peripheral blood smear can reveal schistocytes, though these are rarely seen to exceed 10% of red blood cells (RBCs). The presence of schistocytes is neither sensitive nor specific for DIC, but in certain instances, it may help confirm a chronic DIC diagnosis when the schistocytes are seen in concert with normal coagulation values and increased D-dimer levels.
· Mortality of DIC: 10 – 50%.
· International Society on Thrombosis and Haemostasis (ISTH) score calculation for the laboratory diagnosis of DIC: http://www.cancernetwork.com/sites/default/files/figures_diagrams/1502FeinsteinTable.png
{kind=link}
and
{kind=link}
A Japanese Association for Acute Medicine scoring System of DIC is also useful. For both score systems, see http://emedicine.medscape.com/article/199627-workup#c7
· A multicenter, prospective study concluded that the Japanese Association for Acute Medicine (JAAM) DIC scoring system exhibits good prognostic value in predicting multiple organ dysfunction syndromes (MODS) and poor prognosis in patients with severe sepsis and can detect more patients requiring treatment. Conducting repeated daily JAAM scoring increases the ability to predict the patient’s prognosis (Reference: http://www.ccforum.com/content/pdf/cc12783.pdf ).
· A prospective study concluded that in sepsis, the Japanese Association for Acute Medicine (JAAM) DIC score identified most of the patients diagnosed by the overt International Society on Thrombosis and Haemostasis (ISTH) criteria but failed to discriminate between survivors and non-survivors amongst DIC patients (Reference: http://www.ncbi.nlm.nih.gov/pubmed/22138415 ).
· Chronic DIC: DIC exists in both acute and chronic forms. Acute DIC develops when sudden exposure of blood to procoagulants, such as tissue factor (TF) or tissue thromboplastin, generates intravascular coagulation. Compensatory hemostatic mechanisms are quickly overwhelmed. Consequently, severe consumptive coagulopathy occurs, leading to hemorrhage develops. Abnormalities of blood coagulation parameters are readily identified. Acute DIC frequently results in (multi) organ failure. In contrast, chronic DIC reflects a compensated state that develops when blood is continuously or intermittently exposed to small amounts of TF. Compensatory mechanisms in the liver and bone marrow are not overwhelmed, and there may be little obvious clinical or laboratory indication of the presence of DIC. Chronic DIC is more frequently observed in patients with solid tumors and in those with large aortic aneurysms.
· Differential diagnosis: dysfibrinogenemia, hemolytic–uremic syndrome (HUS) & thrombotic thrombocytopenic purpura (TTP), heparin-induced thrombocytopenia (HIT), idiopathic thrombocytopenic purpura (ITP).
· Treatment: treatment of DIC is centered around treating the underlying condition (DIC can result from numerous clinical conditions, including sepsis, trauma, obstetric emergencies, and malignancy). Transfusions of platelets or fresh frozen plasma (FFP) can be considered in cases of significant bleeding or those with a planned invasive procedure. Platelet and factor replacement should be directed not at simply correcting laboratory abnormalities but at addressing clinically relevant bleeding or meeting procedural needs. The target goal of such transfusion depends on the clinical situation. Cryoprecipitate can be considered in those with a low fibrinogen level. Treatment of thrombosis with anticoagulants such as heparin is rarely used due to the risk of bleeding. Heparin should be provided to those patients who demonstrate extensive fibrin deposition without evidence of substantial hemorrhage; it is usually reserved for cases of chronic DIC. Heparin is appropriate to treat thrombosis that occurs with DIC. It also has limited use in acute hemorrhagic DIC in a patient with a self-limited condition of acral cyanosis and digital ischemia. Antithrombin and antifibrinolytics are generally not used. Studies showed that recombinant human-activated protein C, which was previously recommended in those with severe sepsis and DIC, confers no benefit, so it is not used today. Recombinant factor VII has been proposed by some studies as a ‘last resort’ in those with severe hemorrhage due to obstetric or other causes, but conclusions about its use are still insufficient, and it may also increase thrombosis risk. With this drug, the risk of thrombosis, particularly arterial thromboembolic events (e.g., myocardial ischemia, MI, cerebral ischemia, and/or infarction), may be further increase in non-hemophilic patients who receive factor VIIa (recombinant) for non-FDA-labeled indications. The potentially greater risk in patients with disseminated intravascular coagulation (DIC), advanced atherosclerotic disease, crush injuries, septicemia, or concomitant treatment with activated or nonactivated prothrombin complex concentrates (APCCs or PCCs) due to circulating tissue factor (TF) or predisposing coagulopathy.
NEWBORN – HEMATOLOGICAL FEATURES
· Newborn – hematological features: Decreased Hb and RBCs in the first week of life.
· Common physiological anemia > 48 hours after birth.
· Also, macrocytes (increased MCV) after birth (especially the early 12 hours); 3 – 10 orthochromic normoblasts (erythroid precursors); polychromasia; anisocytosis (15,2 – 18%); occasional spherocytes; higher leukocyte counts; increased segmented and band neutrophils; occasionally a metamyelocyte.
· Lymphocyte morphology is pleomorphic (they range from reactive to mature).
No comments:
Post a Comment