
Dr. James Manos (MD)
January 2, 2016
January 2, 2016
Tips in Hematology
Volume (4)
2nd edition (revised)
CONTENTS
Intermediate or variable: +8, other single or double anomalies
Poor: complex (>3 chromosomal aberrations); chromosome 7 anomalies.
HEMATOLOGICAL MALIGNANCIES
HEMATOLOGICAL MALIGNANCIES – INTRODUCTION
Hematological malignancy investigation/taxonomy
Bone marrow
Dry tap on bone marrow aspiration
Cytochemistry in acute leukemias
ACUTE LEUKEMIAS
Acute myeloid leukemia (AML; also known as acute myelogenous leukemia)
Differential diagnosis between acute lymphoblastic leukemia (ALL) & acute myeloid leukemia (AML)
LYMPHOPROLIFERATIVE DISORDERS (LPDs)
Lymphoproliferative disorders (LPDs)
Αcute lymphoblastic leukemia (ALL; also known as acute lymphocytic leukemia and acute lymphoid leukemia)
Differential diagnosis between acute lymphoblastic leukemia (ALL) & acute myeloid leukemia (AML)
B – cell Chronic lymphocytic leukemia (CLL; also known as chronic lymphoid leukemia)
T – cell Chronic lymphocytic leukemia (CLL; also known as chronic lymphoid leukemia)
Transformation of chronic lymphocytic leukemia (CLL)
B & T – cell prolymphocytic leukemia
Hairy cell leukemia (HCL; also called leukemic reticuloendotheliosis)
Richter’s syndrome (Richter’s transformation) in chronic lymphocytic leukemia (CLL) and hairy cell leukemia (HCL)
MYELODYSPLASTIC SYNDROMES (MDS)
MDS (myelodysplastic syndromes)
MYELOPROLIFERATIVE NEOPLASMS (MPNs)
Myeloproliferative neoplasms (MPNs) disorders
JAK2 mutation (JAK2 V617F test)
Chronic myelogenous leukemia (CML; also called chronic myeloid or myelocytic or myelogenic leukemia and chronic granulocytic leukemia (CGL))
Difference between chronic myelogenous leukemia (CML) and leukemoid reaction
Polycythemia vera (PV, PCV)
Essential thrombocythemia (ET)
Primary myelofibrosis (PMF)
Mastocytosis
PLASMA CELL DYSCRASIAS
Plasma cell dyscrasias
Cryoglobulinemia (CG)
Multiple myeloma (MM)
Monoclonal gammopathy of undetermined significance (MGUS)
Waldenstrom's macroglobulinemia (WM)
LYMPHOMAS
Hodgkin’s lymphoma (HL)
Non-Hodgkin lymphomas (NHLs)
Cutaneous T cell lymphoma (CTCL)
SYNDROMES RELATED TO HEMATOLOGICAL MALIGNANCIES
Sweet's syndrome (SS)
HEMATOLOGICAL MALIGNANCIES
HEMATOLOGICAL MALIGNANCIES – INTRODUCTION
· Hematological malignancy investigation/taxonomy: bone marrow, cytochemistry (e.g., specific esterase positive or negative) & immunophenotyping (e.g., CD 33 positive) & genetical analysis (cytogenetics; chromosomal & molecular, e.g., Ph (Philadelphia) fusion chromosome bcr-abl on CML (chronic myelogenous leukemia)).
· Bone marrow: aspiration (sternum) or trephine histology (biopsy).
· Dry tap on bone marrow aspiration e.g., in myelofibrosis and hairy cell leukemia.
· Α study concluded that the most frequent diagnoses were metastatic carcinoma (17.2%), chronic myelogenous leukemia (14.9%), idiopathic myelofibrosis (13.8%), and hairy cell leukemia (10.3%). The presence of peripheral blood nucleated red blood cells, thrombocytopenia, and elevation of the serum lactate dehydrogenase were frequent findings in patients who experienced dry taps (Reference: http://www.ncbi.nlm.nih.gov/pubmed/2239919 ).
· Cytochemistry in acute leukemias:
· Myeloperoxidase (MPO): present in myeloid cells but not in lymphoid cells; if positive, it rules out ALL
· Νon-specific esterase (NSE): present in large amounts in monocytic cells; positive in AML M4 & AML M5
· Ρeriodic acid Schiff (PAS): block like in some cases of ALL; granular in AML M6
· Αcid phosphatase: dot-like in T-ALL.
ACUTE LEUKEMIAS
· Note: acute lymphoblastic leukemia (ALL) and hairy cell leukemia (HCL) are described on the lymphoproliferative disorders (LPDs)
· Acute myeloid leukemia (AML; also known as acute myelogenous leukemia): the most common form of leukemia in adults. Incidence increases with age.
· It is the most common leukemia in children less than 1 year of age. It is rare in older children and adolescents, but the second peak of incidence occurs among adults aged 40 years.
· Signs & symptoms: anemia (pallor, fatigue, SOB (shortness of breath), decreased platelets (petechia; easy bruising or bleeding with minor trauma), persistent or frequent infections/ fever, etc. LN (lymph node) swelling is rare, contrary to acute lymphoblastic leukemia (ALL). Splenomegaly is typically mild and asymptomatic. Skin involvement 10% (leukemia cutis). Sweet’s syndrome (paraneoplastic inflammation of the skin). Some patients may experience gum swelling. Rarely, a chloroma may occur (a solid leukemic mass or tumor outside the bone marrow). Bone pain and joint pain are seen as the first symptoms in 25% of patients. An enlarged spleen is seen in 50% of all AML patients, but lymph node enlargement is rare.
· Lab tests: FBC/ peripheral blood: the patient usually has an elevated white blood cell count, anemia, thrombocytopenia, and neutropenia. Also, myeloblasts are present. Auer rods: elongated, bluish-red rods composed of fused lysosomal granules, seen in patients with acute myelogenous leukemia (AML).
· Bone marrow: >_20% blasts in bone marrow, total leukocyte count and blasts’ percentage in peripheral blood is variable; cytochemistry (myeloperoxidase MPO (positive on myeloblasts), non–specific esterase (positive on monocytic series); immunophenotyping (CD13 (myeloid), CD33 (myeloid), CD14 (monocytic), Glycophorin A (erythroid), CD41 (megakaryocytic)); specific chromosomal abnormalities in certain types.
· World Health Organization (WHO) classification:
· a) AML with recurrent genetic abnormalities
· b) AML with myelodysplastic changes
· c) Therapy-related myeloid neoplasms
· d) Myeloid sarcoma
· e) Myeloid proliferations related to Down’s syndrome
· f) Blastic plasmacytoid dendritic cell neoplasm
· g) AML not otherwise categorized.
· Treatment: induction and consolidation (post-remission) chemotherapy. Induction chemotherapy in all FAB categories except M3 includes cytarabine and anthracycline (such as daunorubicin), known as the ‘7+3’ or ‘3+7’ scheme.
· Other schemes include high–dose cytarabine alone or FLAG–like regimens [FLudarabine, Ara-C (cytarabine, also called cytosine – arabinoside, G- CSF (granulocyte colony-stimulating factor)].
· AML subtype M3 (APL, acute promyelocytic leukemia) is treated with induction chemo (usually an anthracycline) & all–trans–retinoic acid (ATRA). Its treatment may be complicated by DIC (disseminated intravascular coagulation) when promyelocytes release their granule contents into the peripheral circulation. APL is eminently curable, with well-documented treatment protocols.
· Consolidation treatment is given for good prognosis AML. However, for patients at elevated risk of relapse (e.g., those with high-risk cytogenetics, underlying MDS, or therapy-related AML), allogeneic–stem cell transplantation is usually recommended. For relapsed APL, arsenic trioxide has been tested in trials. The 5–year survival is 70% for the good risk category; 48% for intermediate-risk, and 15% for poor risk. Complete remission is obtained in about 50%–75% of newly diagnosed adults, although this may vary.
· See:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Gum swelling: https://en.wikipedia.org/wiki/File:AMLCase-66.jpg
{kind=link}
· Differential diagnosis between acute lymphoblastic leukemia (ALL) & acute myeloid leukemia (AML):
· Age (ALL more common in children, AML more common in infants & adults)
· Significant lymphadenopathy in > 1 location (common in ALL; uncommon in AML)
· Meningeal disease (more common in ALL; less common in AML)
· Mediastinal lymphadenopathy (seen in T – ALL; rare in AML)
· Size of blasts (small to medium in ALL; large in AML)
· Cytoplasm on blasts (scanty in ALL; moderately abundant in AML)
· Auer rods on blasts (absent in ALL; pathognomic in AML if present)
· Nuclear chromatin on blasts (coarse on ALL; fine on AML)
· Nucleoli on blasts (indistinct, 0 – 2 in ALL; prominent, 1 – 4 in AML)
· MDS (myelodysplasia) (absent in ALL; may be present in AML)
· Cytochemistry: myeloperoxidase MPO (negative in ALL; positive in AML), periodic acid Schiff PAS (block-like positive in 70% of cases in ALL; negative in AML), TdT (terminal deoxynucleotidyl transferase) (positive in ALL; negative in AML)
· CD antigen analysis (B or T cell markers on ALL; myeloid markers (e.g., CD13, CD14, CD15, CD33) on AML).
LYMPHOPROLIFERATIVE DISORDERS (LPDs)
· Lymphoproliferative disorders (LPDs) are conditions in which lymphocytes are produced in excessive quantities; they typically occur in people with compromised immune systems.
· LPS include:
· Follicular lymphoma
· Chronic lymphocytic leukemia (CLL)
· Acute lymphoblastic leukemia (ALL)
· Hairy cell leukemia (HCL)
· B–cell lymphomas
· T–cell lymphomas
· Multiple myeloma
· Waldenstrom’s macroglobulinemia
· Wiskott – Aldrich syndrome
· Post-transplant lymphoproliferative disorder
· Autoimmune lymphoproliferative syndrome (ALPS)
· Lymphoid interstitial pneumonia.
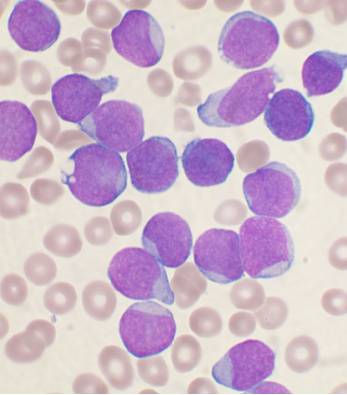
· Αcute lymphoblastic leukemia (ALL; also known as acute lymphocytic leukemia and acute lymphoid leukemia): the most prevalent form of malignancy in childhood (peak 4 – 5 years).
· Usually strikes children between the ages of 2 and 10. The second peak in incidence is seen in elderly patients.
· Signs & symptoms: bone marrow failure (fatigue, infections/ fever, bleeding), organ infiltration (lymphadenopathy, hepatosplenomegaly, CNS (central nervous system) involvement. There is often lymph node enlargement. Enlargement of the spleen (splenomegaly) and of the liver may be seen.
· Lab features: CBC: Neutropenia, thrombocytopenia, and anemia are usually present. Bone marrow: blasts>_ 25% (the marrow specimen is typically hypercellular and characterized by a homogeneous population of cells; leukemia must be suspected in patients whose marrows contain greater than 5%, but the diagnosis should not be made on the basis of a single marrow smear with fewer than 25% blasts), cytochemistry (block-like positivity with PAS (periodic acid Schiff) in some cases of precursor B- ALL; oil red O in ALL – L3)), immunophenotyping, cytogenetic analysis.
· Only 50% of all patients with ALL have increased leukocytes and may not have lymphoblasts in their peripheral blood.
· Classification:
· Ι) Acute lymphoblastic leukemia/lymphoma (former Fab L1/L2).
· a) Precursor B acute lymphoblastic leukemia/ lymphoma.
Cytogenetic subtypes: t(12;21)(p12,q22) TEL/AML-1; t(1;19)(q23;p13) PBX/E2A; t(9;22)(q34;q11) ABL/BCR; T(V,11)(V;q23) V/MLL.
· b) Precursor T acute lymphoblastic leukemia/ lymphoma.
· II) Burkitt’s leukemia/ lymphoma (former FAB L3).
· III) Biphenotyping acute leukemia.
· IV) Variant features:
· 1) Acute lymphoblastic leukemia with cytoplasmic granules.
· 2) Aplastic presentation of ALL.
· 3) Acute lymphoblastic leukemia with eosinophilia.
· 4) Relapse of lymphoblastic leukemia.
· 5) Secondary ALL.
· Treatment: chemotherapy, growth factors, radiation therapy, intensive combined treatments (including bone marrow or stem cell transplantation).
· Chemotherapy:
· a) Remission induction: a combination of prednisone or dexamethasone, vincristine, asparaginase (better tolerated in children) and daunorubicin (adults), CNS (central nervous system) prophylaxis with irradiation, intrathecal cytarabine & MTX (methotrexate) or liposomal cytarabine.
· b) Consolidation/ intensification: cyclophosphamide, cytarabine, daunorubicin, etoposide, thioguanine or mercaptopurine in different combinations. CMS protection with intrathecal MTX (methotrexate) or cytarabine, +_ craniospinal irradiation. Treatment of CNS involvement with intrathecal hydrocortisone, MTX & cytarabine.
· c) Maintenance therapy: mercaptopurine (oral, daily), MTX (oral, once weekly), vincristine (IV once monthly, 5-day course), corticosteroids (oral).
· Immunotherapy: Chimeric Antigen Receptors (CAR): engineered receptors, which graft an arbitrary specificity onto an immune effector cell. These receptors are used to graft the specificity of a monoclonal antibody onto a T cell, with the transfer of their coding sequence facilitated by retroviral vectors. This method, on ALL, uses a single-chain variable fragment (scFv) designed to recognize the cell-surface marker CD19 to treat ALL. CD19 is a molecule found on all B-cells and can be used to distinguish the potentially malignant B-cell population in the patient. In this therapy, mice are immunized with the CD19 antigen, thereby producing anti-CD19 antibodies.
· Prognosis: The 5-year survival rate has improved to 85% due to clinical trials of new therapeutic agents and improvements in stem cell transplantation. A cure is achieved in over 80% of affected children, although only 20 – 40% of adults can be cured.
· See:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
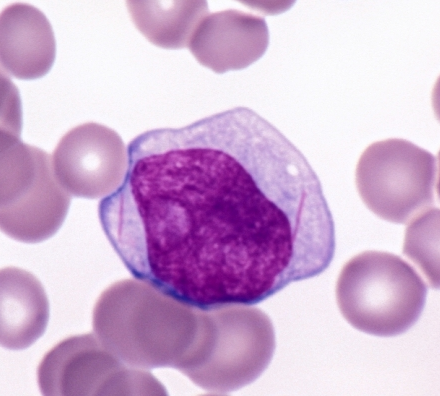
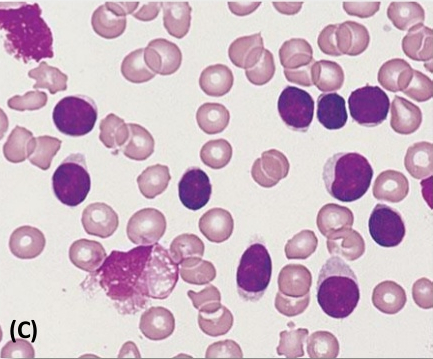
· B – cell Chronic lymphocytic leukemia (CLL, also known as chronic lymphoid leukemia): is the most common type of leukemia. The disease is usually discovered when other medical problems are present.
· A neoplastic proliferation of mature-looking, functionally incompetent B lymphocytes.
· Usually occurs in older patients; it is rare in patients less than 40 years of age. Mean Age > 50 years.
· Signs & symptoms: weakness, fatigue, weight loss, generalized lymphadenopathy, splenomegaly; 25% asymptomatic.
· Lab features – CBC/ Blood smear: The malignant cell in CLL is usually a small, mature-appearing lymphocyte.
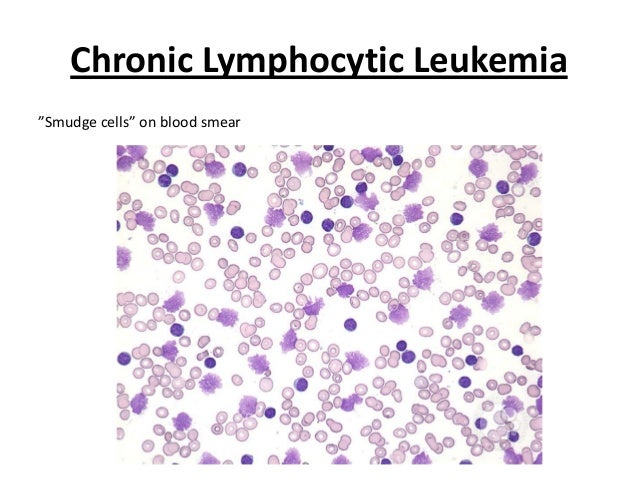
· Absolute lymphocytosis (> 5,000/cmm), neoplastic lymphocytes are small, mature-looking, with dense clumped chromatin and scanty cytoplasm; smudge cells are also characteristic.
· Smudge cells are leukocytes that have been damaged during the preparation of the peripheral blood smear. This usually occurs due to cellular fragility. They are usually seen in chronic lymphocytic leukemia (CLL). However, they may be an artifact of slide preparation. A study concluded that the percentage of smudge cells on a blood smear is readily available and an independent factor predicting overall survival in CLL (Reference: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2668708/ )
· Immunophenotyping: CD19+, CD20 + (weak), CD5+, CD23 +, weak surface membrane Ig+, a single light chain, FMC7 –, CD2 –. In CLL, the lymphocytes of the B cell lineage are genetically clonal, expressing CD19 and CD20 and characteristically CD5 and CD23.
· Rai Staging System:
· Stage 0: absolute lymphocytosis (>15 000/mm3) without adenopathy, hepatosplenomegaly, anemia, or thrombocytopenia.
· Stage I: absolute lymphocytosis with lymphadenopathy without hepatosplenomegaly, anemia, or thrombocytopenia.
· Stage II: absolute lymphocytosis with either hepatomegaly or splenomegaly with or without lymphadenopathy.
· Stage III: characterized by absolute lymphocytosis and anemia (hemoglobin <11 g/dL) with or without lymphadenopathy, hepatomegaly, or splenomegaly.
· Stage IV: characterized by absolute lymphocytosis and thrombocytopenia (<100 000/mm3) with or without lymphadenopathy, hepatomegaly, splenomegaly, or anemia.
· Binet Classification:
· Clinical stage A: no anemia or thrombocytopenia and also fewer than three areas of lymphoid involvement (Rai stages 0, I, and II).
· Clinical stage B: no anemia or thrombocytopenia and also with three or more areas of lymphoid involvement (Rai stages I and II).
· Clinical stage C: anemia and/or thrombocytopenia regardless of the number of areas of lymphoid enlargement (Rai stages III and IV).
· Treatment: CLL is treated by chemotherapy, radiation therapy, biological therapy (e.g., in one trial, scientists used genetically modified T cells to attack cells that expressed CD19), or bone marrow transplantation. Symptoms are sometimes treated surgically, such as splenectomy on splenomegaly or by radiation therapy for "debulking" swollen lymph nodes.
· Combination chemotherapy regimens are effective in both newly diagnosed and relapsed CLL. Combinations of fludarabine with alkylating agents (cyclophosphamide) produce higher response rates and longer progression-free survival than single agents: FC (fludarabine & cyclophosphamide), FR (fludarabine & rituximab), FCR (fludarabine, cyclophosphamide & rituximab), CHOP (cyclophosphamide, doxorubicin, vincristine & prednisolone).
· Immunotherapy with monoclonal antibodies is also used: e.g., alemtuzumab (directed against CD52), rituximab, and ofatumumab (directed against CD20). New agents include ibrutinib (a BTK (Bruton’s tyrosine kinase) inhibitor) and idelalisib (a PI3K (phosphatidylinositide 3-kinase) inhibitor that targets the PI3Kδ pathway).
· Prognosis: for all stages of CLL, on average, 44% of men and 52% of women will live for at least 5 years after being diagnosed. However, the personal outlook depends on the stage at which someone is diagnosed. In the earliest stage (A), people generally live for 10 years or more. People diagnosed in the middle stage (B) generally live for around 5 to 8 years. People diagnosed in the most advanced stage (C) usually live for around 1 to 3 years.
· See:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Smudge cells:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· T–cell chronic lymphocytic leukemia (CLL; also known as chronic lymphoid leukemia): chronic T-cell lymphocytic leukemia is a subtype of CLL (chronic lymphocytic leukemia). The subtypes of T-cell leukemia include:
· Large granular lymphocytic leukemia (LGLL). The cytotoxic response to a specific antigen is mediated by two highly professional, although extremely different players, cytotoxic T lymphocytes and natural killer (NK) cells, with the common feature of a morphological appearance of large granular lymphocytes (LGL): both are lymphocytes with coarse azurophilic granules within their cytoplasm, containing the cytotoxic weapons they are equipped with. Clonal expansions of LGL may comprise both T- and NK-cell-derived diseases, but the WHO considers NK-cell-derived diseases as a separate group referred to as chronic lymphoid disorders of NK cells. T-cell LGL (T-LGL) leukemia represents the clonal expansion of a terminally differentiated cytotoxic T-cell bearing a fully functional α/β+ T-cell receptor (TCR) with the corresponding CD8+ CD4− or, rarely, CD4+ CD8−/+dim phenotypic pattern; expansions of γ/δ+ TCR cells account for only rare cases. Less information is available for NK cells in chronic lymphoproliferative disorders (CLPD), although it seems that these cells are activated in vivo by persistent/active infection/stimulation.
· LGLL is a slow-growing T-cell leukemia and is more common in women than in men. The cause of LGLL is unknown, although about 30% of people with LGLL also have rheumatoid arthritis (RA).
See
See
{kind=link}
{kind=link}
{kind=link}
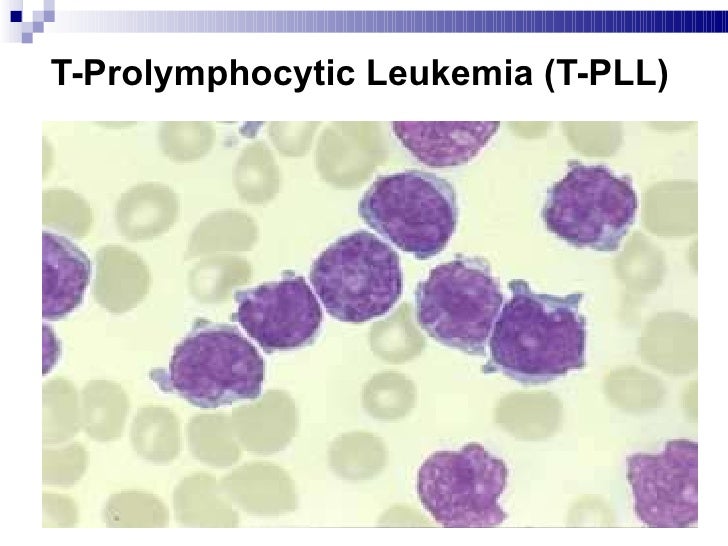
· T-cell prolymphocytic leukemia (T-PLL). T-PLL is an aggressive subtype of CLL. It is more common in older men, but women may also develop T-PLL. It can affect the skin but in a different way than Sezary syndrome (see below).
See
See
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Adult T-cell leukemia/lymphoma (ATLL). ATLL has four subtypes. Depending on the features, it is subclassified as smoldering, chronic, or acute, or as adult T-cell lymphoma, which is a cancer of the lymph system. Acute and adult T-cell lymphoma subtypes grow rapidly. ATLL is caused by a retrovirus called the human T-cell leukemia virus (HTLV1).
See http://o.quizlet.com/ZqY50CwOYKY0O59QAbMx2g_m.png and http://imagebank.hematology.org/Content%5C11861%5C11863%5C11863_normal.J
See http://o.quizlet.com/ZqY50CwOYKY0O59QAbMx2g_m.png and http://imagebank.hematology.org/Content%5C11861%5C11863%5C11863_normal.J
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Flower-like lymphocytes: Peripheral blood smear may show flower-like lymphocytes. This may be from Adult T-cell Leukemia/Lymphoma (ATLL; it is caused by a retrovirus called the human T-cell leukemia virus (HTLV1). However, evaluation of flower-like lymphocytes must be performed carefully because these cells are not only found in ATLL patients but also in patients with various other diseases involving reactive or neoplastic lesions, or even in healthy individuals. They may be found in the peripheral blood in routine daily blood examination, such as in a hepatitis B or anemia patient. Flower-like lymphocytes should also be considered for the differential diagnosis of flower-like plasma cells. In Sezary syndrome, lymphocytes may have a flower-like or a cerebriform nuclear shape in the peripheral blood. For flower-like lymphocytes, see https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9160704/ and https://imagebank.hematology.org/image/12563/flower-cells-of-leukemia
· Cutaneous T cell lymphoma (CTCL): a non-Hodgkin lymphoma class. Unlike most non-Hodgkin lymphomas, which are generally B-cell-related, CTCL is caused by a mutation in T cells. The malignant T cells in the body initially migrate to the skin, causing lesions that change shape as the disease progresses, typically beginning as an itchy rash and eventually forming plaques and tumors before metastasizing to other parts of the body.
· Of all cancers involving the same class of blood cells, 2% of cases are cutaneous T-cell lymphomas. There is some evidence of a relationship with HTLV (human T-lymphotropic virus).
· Classification: cutaneous T-cell lymphomas may be divided into the following types:
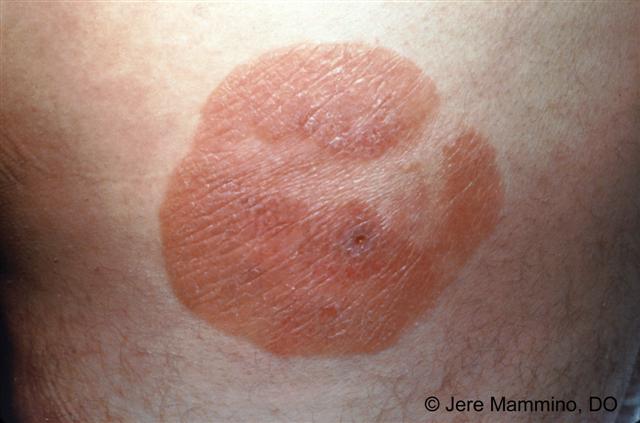
· Mycosis fungoides
{kind=link}
{kind=link}
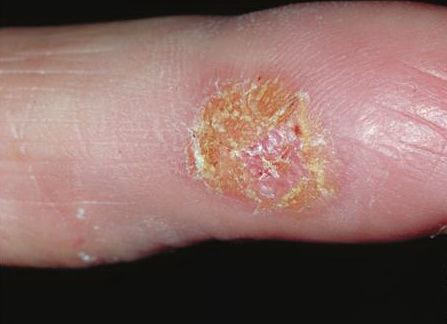
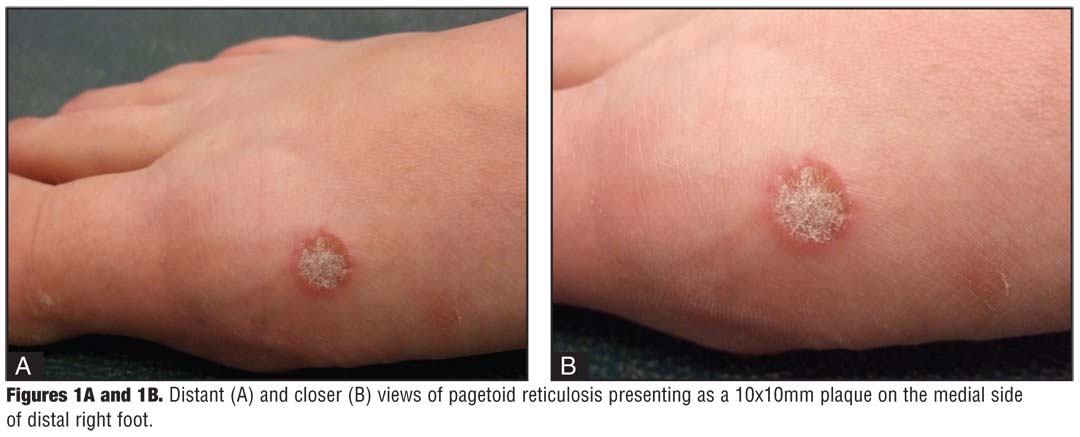
· Pagetoid reticulosis
{kind=link}
{kind=link}
· Sezary syndrome
{kind=link}
{kind=link}
· Granulomatous slack skin
· Lymphomatoid papulosis
· Pituriasis lichenoides chronica
· Pityriasis lichenoides et varioliformis acuta
· CD30+ cutaneous CD30+ large lymphoma
· Non – mycosis fungoides CD30 – cutaneous large T – cell lymphoma
· Pleomorphic T–cell lymphoma
{kind=link}
{kind=link}
· Lannert Lymphoma
· Subcutaneous T–cell lymphoma
· Angiocentric lymphoma
· Blastic NK–cell lymphoma.
· Sezary syndrome. Sezary syndrome is a form of mycosis fungoides, a T-cell lymphoma that occurs in the skin. Sezary syndrome is usually slow-growing and takes years to develop from mycosis fungoides, which is limited to the skin. Sezary syndrome is generally diagnosed when large numbers of lymphoma cells are found in the blood, often together with erythroderma (reddening of the skin). In Sezary syndrome, lymphocytes may have a flower-like or a cerebriform nuclear shape in the peripheral blood.
· Sezary syndrome:
{kind=link}
{kind=link}
1338786565528.jpg){kind=link}
· Mycosis fungoides:
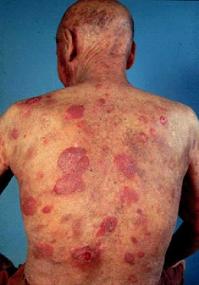{kind=link}
{kind=link}
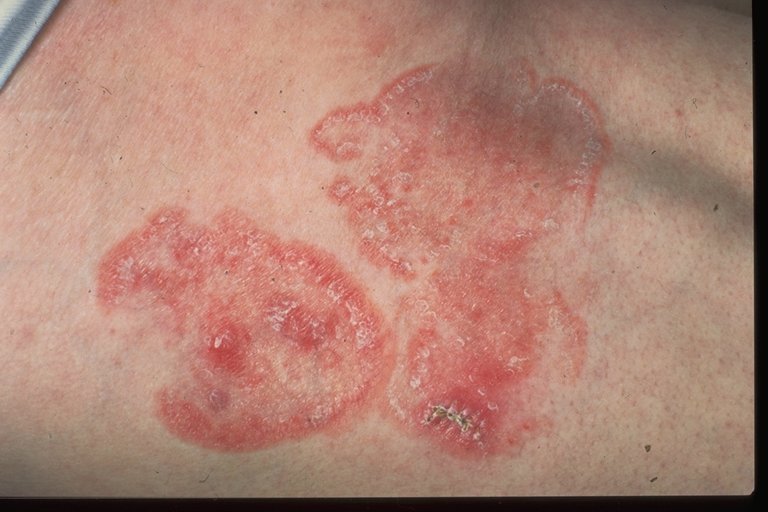{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Transformation of chronic lymphocytic leukemia (CLL):
· Chronic lymphocytic leukemia (CLL) is an indolent B-cell non-Hodgkin's lymphoma that may transform into higher-grade lymphoma. The transformation involves an increased number of prolymphocytic cells, termed prolymphocytic transformation (PLT) or the development of diffuse large B-cell lymphoma (DLBL), also referred to as Richter's transformation (RT).
· A study concluded that in the case of large B-cell lymphoma, generally thought to arise from the chronic lymphocytic leukemia clone, approximately one-half of the patients had genetically unrelated cancers. In prolymphocytic transformation, all cases studied appeared to evolve from the chronic lymphocytic leukemia clone. The few studies of acute lymphoblastic leukemia and multiple myeloma showed genetic relatedness in some instances and unrelatedness in others. These data indicate that progression to more aggressive B-cell cancers in persons with chronic lymphocytic leukemia can result from clonal evolution or an independent transforming event (Reference: http://annals.org/article.aspx?articleid=706496 ).
· B & T – cell prolymphocytic leukemia (PLL):
· Note: Prolymphocytic leukemia may be caused by the transformation of CLL (chronic lymphocytic leukemia). It can be classified as a kind of CLL.
· B-cell prolymphocytic leukemia is a more aggressive form of leukemia. The malignant B-cells are larger than average. Chromosomal anomalies included deletions from chromosomes 11 & 13. It has been suggested that some cases may represent a variant of mantle cell lymphoma. It has a relatively poor prognosis. Splenic irradiation has been used in the treatment.
· T-cell-prolymphocytic leukemia (T-PLL) is a mature T–cell leukemia with aggressive behavior and predilection for blood, bone marrow, lymph nodes, liver, spleen & skin involvement. It is a very rare leukemia, primarily affecting adults over the age of 30. It represents 2% of all small lymphocytic leukemias in adults.
· Patients typically have a systemic disease at presentation, including hepatosplenomegaly, generalized lymphadenopathy, and skin infiltrates.
· Leukemic cells can be found in peripheral blood, lymph nodes, bone marrow, spleen, liver, and skin.
· FBC (CBC): A high lymphocyte count (> 100 x 109/L) along with anemia & thrombocytopenia are common findings.
· HTLV – 1 serology is negative.
· In the peripheral blood, T-PLL consists of medium-sized lymphocytes with single nucleoli and basophilic cytoplasm with occasional blebs or projections. The nuclei are usually round to oval, with occasional patients having cells with a more irregular nuclear outline similar to the cerebriform nuclear shape seen in Sezary syndrome. A small cell variant comprises 20% of all T-PLL cases, and the Sézary cell-like (cerebriform) variant is seen in 5% of cases.
· Marrow involvement is typically diffuse with morphology similar to what is observed in peripheral blood.
· Treatment: T-cell prolymphocytic leukemia is difficult to treat and does not respond to most available chemotherapeutic drugs. Many different treatments have been used, with limited success: purine analogs (pentostatin, fludarabine, cladribine), chlorambucil, and various forms of combination chemotherapy, including cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), cyclophosphamide, vincristine, prednisone (COP), and vincristine, doxorubicin, prednisone, etoposide, cyclophosphamide, bleomycin (VAPEC-B). Alemtuzumab is an anti–CD52 monoclonal antibody that attacks white blood cells and has been used in treatment with greater success. In one study of previously treated people with T-PLL, people who had a complete response to alemtuzumab survived a median of 16 months after treatment. Some patients who successfully respond to treatment also undergo stem cell transplantation to consolidate the response.
· Prognosis: T-PLL is an aggressive disease. Before the recent introduction of improved treatments, such as alemtuzumab, the median survival time after diagnosis was 7.5 months. Recently, some patients have survived for 5 years or more, although the median survival remains low.
· Images:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Hairy cell leukemia (HCL; also called leukemic reticuloendotheliosis): a rare malignant disorder that usually occurs in middle-aged patients over 50.
· A rare chronic lymphoproliferative disorder of B-cell origin.
· Usually, in middle-aged persons.
· HCL is usually classified as a subtype of CLL and makes up approximately 2% of all leukemias.
· Signs & symptoms: the first symptoms of the disease include weakness and lethargy. Also, tiredness, easy bruising, recurrent infections, splenomegaly (80–90%), and peripheral lymphadenopathy in less than 5% of patients, but abdominal lymphadenopathy is a relatively common finding on CT scans. In some patients, it may be detected incidentally on a peripheral blood smear.
· Lab features/ CBC/ peripheral blood smear: cytopenia, hairy cells in bone marrow & peripheral blood. Bone marrow: dry tap; infiltration by hairy cells (honeycomb appearance on biopsy), increased marrow fibrosis.
· Hairy cells are characterized by their fine, irregular pseudopods and immature nuclear features.
· Bone marrow aspiration is often unsuccessful (dry tap) because of complete infiltration by hairy cells, resulting in a dispersed spongy web of cells in an increased meshwork of reticulin fiber.
· Immunochemistry: hairy cells positive for TRAP (tartrate-resistant acid phosphatase).
· Immunophenotyping: CD19+. CD11c+, CD25+, SmIg (surface membrane immunoglobulin)+ (strong), CD103+, HC2+, CD5. Hairy cell leukemia-variant (HCL-V), which shares some characteristics with B cell prolymphocytic leukemia (B-PLL), does not show CD25.
· Variants: Two variants have been described:
· a) Hairy cell leukemia-variant (HCL – V) is usually diagnosed in men.
· b) Japanese variant.
· The non-Japanese variant is more difficult to treat than classical HCL and HCL-V. Hairy cell leukemia-Japanese variant (HCL-J) is more easily treated.
· Hairy cell leukemia-variant (HCL-V) is a more aggressive disease. It is less likely to be treated successfully than classic HCL, and remissions tend to be shorter.
· HCL-V differs from classic HCL in the following:
· a) Higher WBCs (sometimes > 100 000 cells/ microliter)
· b) A more aggressive course of disease requiring more frequent treatment
· c) Hairy cells have an unusually large nucleolus for their size
· d) Production of little excess fibronectin produced by classic hairy cells
· e) Interfere with bone marrow biopsies
· f) Low or no cell-surface expression of CD25 (also called Interleukin – 2 receptor alpha chain, or p55) that (these low levels) may explain why HCL-V cases are generally much more resistant to treatment by immune system hormones.
· Treatment of HCL: chemotherapy (purine analogs; cladribine (2CDA) and pentostatin (DCF)).
· Immunotherapy: if a patient is resistant to either cladribine or pentostatin; monoclonal antibodies that destroy cancerous B cells, such as rituximab, alemtuzumab, ibritumomab, tiuxetan, I-131, and also tositumomab for refractory cases.
· Interferon-alpha (in about 65% of patients, the drug helps stabilize the disease or produce a slow, minor improvement for a partial response)
· Splenectomy (on splenomegaly)
· Bone marrow transplant.
· Treatment of HCL–V: combination chemotherapy regimen "CHOP," interferon-alpha, and common alkylating agents such as cyclophosphamide provide minimal benefit. Pentostatin and cladribine provide some benefit to many HCL-V patients but typically induce shorter remission periods and lower response rates than when they are used in classic HCL. More than half of patients respond partially to splenectomy.
· Hairy cell leukemia-Japanese variant (HCL-J) is more easily treated, e.g., with cladribine.
· Prognosis: more than 95% of new patients are treated well/ adequately by cladribine or pentostatin. Most new patients can expect a disease-free remission span of about ten years, or sometimes much longer, after taking one of these drugs just once. With appropriate treatment, the overall projected lifespan for patients is normal or near-normal.
· In all patients, the first two years after diagnosis have the highest risk for a fatal outcome; HCL-V patients routinely survive for more than 10 years, and younger patients can likely expect a long life.
· See:
· http://www.ijpmonline.org/articles/2012/55/1/images/IndianJPatholMicrobiol_2012_55_1_61_94858_f1.jpg
{kind=link}
{kind=link}
![http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/hairy_cell_leukemia_classic/hairy-cell-leukemia-[7-bl089-3].jpeg?Width=600&Height=450&Format=4](http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/hairy_cell_leukemia_classic/hairy-cell-leukemia-%5b7-bl089-3%5d.jpeg?Width=600&Height=450&Format=4){kind=link}
![http://www.pathpedia.com/education/eatlas/histopathology/bone_marrow/hairy_cell_leukemia_bone_marrow/hairy-cell-leukemia-[9-bm083-9].jpeg?Width=600&Height=450&Format=4](http://www.pathpedia.com/education/eatlas/histopathology/bone_marrow/hairy_cell_leukemia_bone_marrow/hairy-cell-leukemia-%5b9-bm083-9%5d.jpeg?Width=600&Height=450&Format=4){kind=link}
{kind=link}
· Hairy cell leukemia-variant (HCL – V):
{kind=link}
{kind=link}
{kind=link}
· Richter’s syndrome (Richter’s transformation) in Chronic lymphocytic leukemia (CLL) and hairy cell leukemia (HCL): Richter's syndrome (RS), also known as Richter's transformation, is a transformation that occurs in about 5 – 10% of B cell chronic lymphocytic leukemia (CLL) and hairy cell leukemia into a fast-growing diffuse large B cell lymphoma (a variety of non-Hodgkin lymphoma) which is refractory to treatment and has a bad prognosis. There is also a less common variant in which CLL can progress to Hodgkin lymphoma. Rarely, transformations to myeloid leukemia may occur. Richter's transformation affects about 5% of CLL patients at some point during their lives. The prognosis is generally poor. The Richter syndrome (RS) score, which estimates prognosis, is based on the patient's performance status, LDH, platelet count, lymphoma tumor size, and number of prior therapies received. Overall, the median survival is between five to eight months. The Hodgkin's lymphoma variant of Richter's carries a better prognosis than the predominant diffuse large B-cell lymphoma type, but worse than a de novo case of Hodgkin's.
{kind=link}
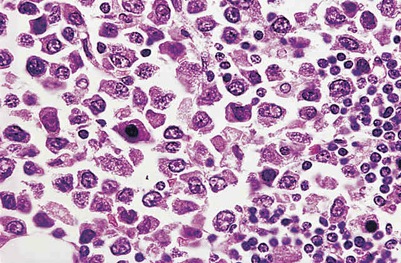{kind=link}
MYELODYSPLASTIC SYNDROMES (MDS)
· MDS (myelodysplastic syndromes): acquired clonal stem disorders with dysplasia of 1 or more hematopoietic cell lines, ineffective erythropoiesis, and increased risk of transformation to AML (acute myelogenous leukemia),
· The median age at diagnosis of an MDS is between 60 and 75 years; a few patients are younger than fifty; rare in children. Males are slightly more commonly affected than females.
· Incidence: the incidence is probably increasing as the age of the population increases, and some authors propose that the incidence in patients over 70 may be as high as 15 cases per 100,000 annually.
· Signs & symptoms: many people are asymptomatic, and blood cytopenia or other problems are identified as part of a routine blood count: cytopenia: anemia (fatigue, weakness), neutropenia (fever, infections), thrombocytopenia (bleeding tendencies). Also, splenomegaly, but rarely hepatomegaly.
· Chromosomal abnormalities.
· FBC/ peripheral blood smear: cytopaenia of 1 or more cell lines, oval macrocytosis, dimorphic RBC population (e.g., the presence of macrocytic & normocytic RBCs), pseudo–Pelger Huët neutrophils (hypolobated), hypoglanular neutrophils, giant platelets.
· Bone marrow: features of dyserythropoiesis and dysmegakaryopoiesis, abnormal localization of immature precursors, ringed sideroblasts in some cases. Cytogenetic abnormalities.
· Classification (WHO):
· i) Refractory cytopenia with unilineage dysplasia (RCUD).
· ii) Refractory anemia with ringed sideroblasts (RARS).
· iii) Refractory cytopenia with multilineage dysplasia (RCMD).
· iv) Refractory anemia with excess blasts-1 (RAEB-1).
· v) Refractory anemia with excess blasts-2 (RAEB-2).
· vi) Myelodysplastic syndrome, unclassified (MDS-U).
· vii) Myelodysplastic syndrome associated with isolated del(5q).
· Prognosis: although there is a risk of developing AML, about 50% of deaths occur as a result of bleeding or infection. Leukemia arising from myelodysplasia is notoriously resistant to treatment. The median survival rate varies from years to months, depending on the type. Stem cell transplantation offers survival rates of 50% at 3 years, although older patients do poorly.
· Indicators of a good prognosis: younger age; normal or moderately reduced neutrophil or platelet counts; low blast counts in the bone marrow (<20%) and no blasts in the blood; no Auer rods; ringed sideroblasts; normal karyotypes or mixed karyotypes without complex chromosome abnormalities and in vitro marrow culture: non-leukemic growth pattern.
· Indicators of a poor prognosis: advanced age; severe neutropenia or thrombocytopenia; high blast count in the bone marrow (20 – 29%) or blasts in the blood; Auer rods; absence of ringed sideroblasts; abnormal localization of immature granulocyte precursors in bone marrow section all or mostly abnormal karyotypes or complex marrow chromosome abnormalities and in vitro bone marrow culture-leukemic growth pattern.
· Prognosis and karyotype: Good: Normal, -Y, del(5q), del(20q)
· The International Prognosis Scoring System (IPSS) is commonly used to predict long-term outcomes. For a calculator of the IPPS, see: http://www.mds-foundation.org/ipss-r-calculator/
· Treatment: The IPSS scoring system can help triage patients for more aggressive treatment, such as bone marrow transplant, and help determine the best timing of this therapy. Supportive care with blood product support and hematopoietic growth factors, such as erythropoietin (EPO), is the mainstay of treatment. Three agents have been approved by the FDA of the USS for the treatment of MDS: 5 – 5-azacytidine (21-month median survival), decitabine (complete response rate has been reported as high as 43%; a phase I study has shown efficacy in AML when decitabine is combined with valproic acid, an antiepileptic), lenalidomide (effective in reducing red blood cell transfusion requirement in patients with the chromosome 5q deletion subtype of MDS). Clinical trials with iron-chelating agents have demonstrated the reversal of some of the consequences of iron overload in MDS.
MYELOPROLIFERATIVE NEOPLASMS (MPNs)
· Myeloproliferative neoplasms (MPNs) disorders: a group of diseases of the bone marrow in which excess cells are produced. They are related to and may evolve into MDS (myelodysplastic syndrome) and AML (acute myelogenous leukemia), although the myeloproliferative diseases, overall, have a much better prognosis than these conditions.
· Classification of MPNs (WHO):
· a) CML [chronic myelogenous leukemia; it is Philadelphia (Ph) chromosome (oncogenic bcr – abl fusion gene) positive; the rest are Ph chromosome-negative
· b) Polycythemia vera (PV)
· c) Essential thrombocythemia
· d) Primary myelofibrosis
· e) Chronic neutrophilic leukemia
· f) Chronic eosinophilic leukemia (not otherwise specified)
· g) Μastocytosis.
· Cytogenetics: most Philadelphia chromosome-negative cases have an activating JAK2 or MPL mutation. Mutations in CALR have been found in the majority of JAK2- and MPL-negative cases of essential thrombocythemia and myelofibrosis.
· Further classification:
· a) Chronic myelogenous leukemia (CML), with defining translocation t(9;22); Philadelphia chromosome, BCR-ABL translocation, which has three breakpoints: i) u-BCR-ABL (p230) (it leads to CML with usual neutrophilia and basophilia). ii) Minor-BCR-ABL (p190) (it leads to CML, which tends to become acute lymphoblastic leukemia (ALL), usually precursor B–ALL and rarely precursor T-ALL). iii) Major-BCR-ABL (p210): usual normal breakpoint.
· b) Essential thrombocythemia (ET), associated with the JAK2V617F mutation in up to 55% of cases and with an MPL (thrombopoietin receptor) mutation in up to 5% of cases. Cellular phase and fibrotic phase.
· c) Polycythemia vera (PV), associated most often with the JAK2V617F mutation in more than 95% of cases, whereas the remainder has a JAK2 exon 12 mutation. Cellular and fibrotic phase.
· d) Primary myelofibrosis (PMF), associated with the JAK2V617F mutation in up to 50% of cases, the JAK2 exon 12 mutations in 1 – 2% of cases, and the MPL (thrombopoietin receptor) mutation in up to 5% of cases. Cellular and fibrotic phase.
· Calreticulin (CALR) mutations have been reported in Janus kinase 2 (JAK2) and myeloproliferative leukemia (MPL) – negative essential thrombocythemia and primary myelofibrosis. A study demonstrated that CALR mutations can also be associated with JAK2-negative PV (Reference: http://www.ncbi.nlm.nih.gov/pubmed/25305205 ).
· Treatment: no curative drug treatment exists for any of the MPDs. The goal of treatment for ET and PV is the prevention of thrombo-hemorrhagic complications, and the goal of treatment for MF is the amelioration of anemia, splenomegaly, and other symptoms. Low-dose aspirin is effective in PV and ET. Tyrosine kinase inhibitors, such as imatinib, have improved the prognosis of CML patients. A JAK2 inhibitor (ruxolitinib) has been approved for use in primary myelofibrosis, and trials of these inhibitors are in progress for the treatment of other myeloproliferative neoplasms such as polycythemia vera.
· JAK2 mutation (JAK2 V617F test) (blood, bone marrow): the JAK2 V617F test may be ordered along with other tests when a health care provider suspects that a person has a blood disorder known as a myeloproliferative neoplasm (MPN), especially polycythemia vera (PV), essential thrombocythemia (ET), or primary myelofibrosis (PMF). Somatic JAK2 gene mutations are also associated with several related conditions.
· The V617F mutation is occasionally found in people with leukemia or other bone marrow disorders. Budd-Chiari syndrome, which results from a blocked hepatic vein in the liver, can also be associated with the V617F mutation when it is caused by an underlying bone marrow disorder. It is unknown how one particular mutation can be associated with several conditions.
· Note: the JAK2 exon 12 test may be ordered when the JAK2 V617F test is negative and the doctor still suspects polycythemia vera (PV).
· Chronic myelogenous leukemia (CML; also called chronic myeloid or myelocytic or myelogenic leukemia and chronic granulocytic leukemia (CGL)): it is one of the myeloproliferative disorders; characterized by the predominant proliferation of granulocytic cells in the bone marrow; the presence of the Philadelphia chromosome (the result of a reciprocal translocation between chromosome 9 and chromosome 22, which is specifically designated t(9;22)(q34;q11); it gives rise to a fusion gene, bcr-abl, that juxtaposes the Abl1 gene on chromosome 9 – region q34, to a part of the BCR – "breakpoint cluster region" – gene on chromosome 22, region q11).
· About 90% of the patients are diagnosed during the chronic stage, which is most often asymptomatic. In these cases, it may be diagnosed incidentally when an elevated WBC (white blood cell) count is found on a routine lab test. It can also present with symptoms indicative of an enlarged spleen and liver, and the resulting upper-quadrant pain.
· Can occur at any age but is most common after the age of 45 years. The median age at diagnosis is the 5th & 6th decades.
· Stages: 3 stages: chronic (indolent phase, lasts for an average of 3 years from diagnosis), accelerated (increasing loss of differentiation of neoplastic cells; the average duration is 6 months), and blastic stage (transformation to acute leukemia).
· Accelerated phase (WHO criteria): diagnosed any of the following are present on a patient with CML: 10 – 19% myeloblasts in the blood or bone marrow; > 20% basophils in the blood or bone marrow; platelet count < 100 000, unrelated to therapy; platelet count > 100 000, unresponsive to therapy; cytogenetic evolution with new abnormalities in addition to the Philadelphia chromosome; increasing splenomegaly or white blood cell count, unresponsive to therapy.
· Blast crisis (the final phase in the evolution of CML; it behaves like acute leukemia with rapid progression and short survival): diagnosed if any of the following are present in a patient with CML: >20% myeloblasts or lymphoblasts in the blood or bone marrow; large clusters of blasts in the bone marrow biopsy; development of a chloroma (the solid focus of leukemia outside the bone marrow).
· Clinical signs/symptoms: weakness, weight loss, splenomegaly.
· CBC/ peripheral blood smear: there is anemia, markedly elevated levels of leukocytes, thrombocytosis, eosinophilia, basophilia, and a predominance of myelocytes in the peripheral blood. Myeloblasts constitute fewer than 10% of circulating leukocytes. Occasional nucleated red blood cells are seen.
· Neutrophils, metamyelocytes & myelocytes predominate; basophils are increased.
· Βone marrow: hypercellular; marked increased myeloid to erythroid ratio; blasts < 10%.
· Leukocyte alkaline phosphatase (LAP score) activity: abnormally low or absent.
· Cytogenetics: Philadelphia chromosome.
· Molecular genetic: rearrangement of bcr/abl (demonstrated by fluorescent in situ hybridization (IFU)).
· Treatment: The only curative treatment for CML is a bone marrow transplant or an allogeneic stem cell transplant. Chemotherapy, interferon, and steroids used to treat CML in the chronic phase have been replaced by BCR–ABL tyrosine kinase inhibitors, medications that specifically target BCR-ABL, the constitutively active tyrosine kinase fusion protein caused by the Philadelphia chromosome translocation (see above). The first of this new class of drugs was imatinib (Gleevec (R) or Glivec (R)), which was found to inhibit the progression of CML in most patients (65 – 75%). In 2011, a follow-up of 832 patients with imatinib who achieved a stable cytogenetic response reported an overall survival rate of 95.2% at 8 years, similar to the general population rate. Less than 1% of patients died because of leukemia progression. Two novel agents were developed to overcome imatinib resistance and increase responsiveness to TK inhibitors. The first, dasatinib, blocks several further oncogenic proteins, in addition to more potent inhibition of the BCR-ABL protein. A second new TK inhibitor, nilotinib, has also been approved. In 2010, nilotinib and dasatinib were also approved for first-line therapy.
· Note: the presence of the Philadelphia (Ph) chromosome is not sufficiently specific to diagnose CML since it is also found in ALL (acute lymphoblastic leukemia) (25 – 30% in adults and 2 – 10% in pediatric cases) and occasionally in AML (acute myeloid leukemia).
· See:
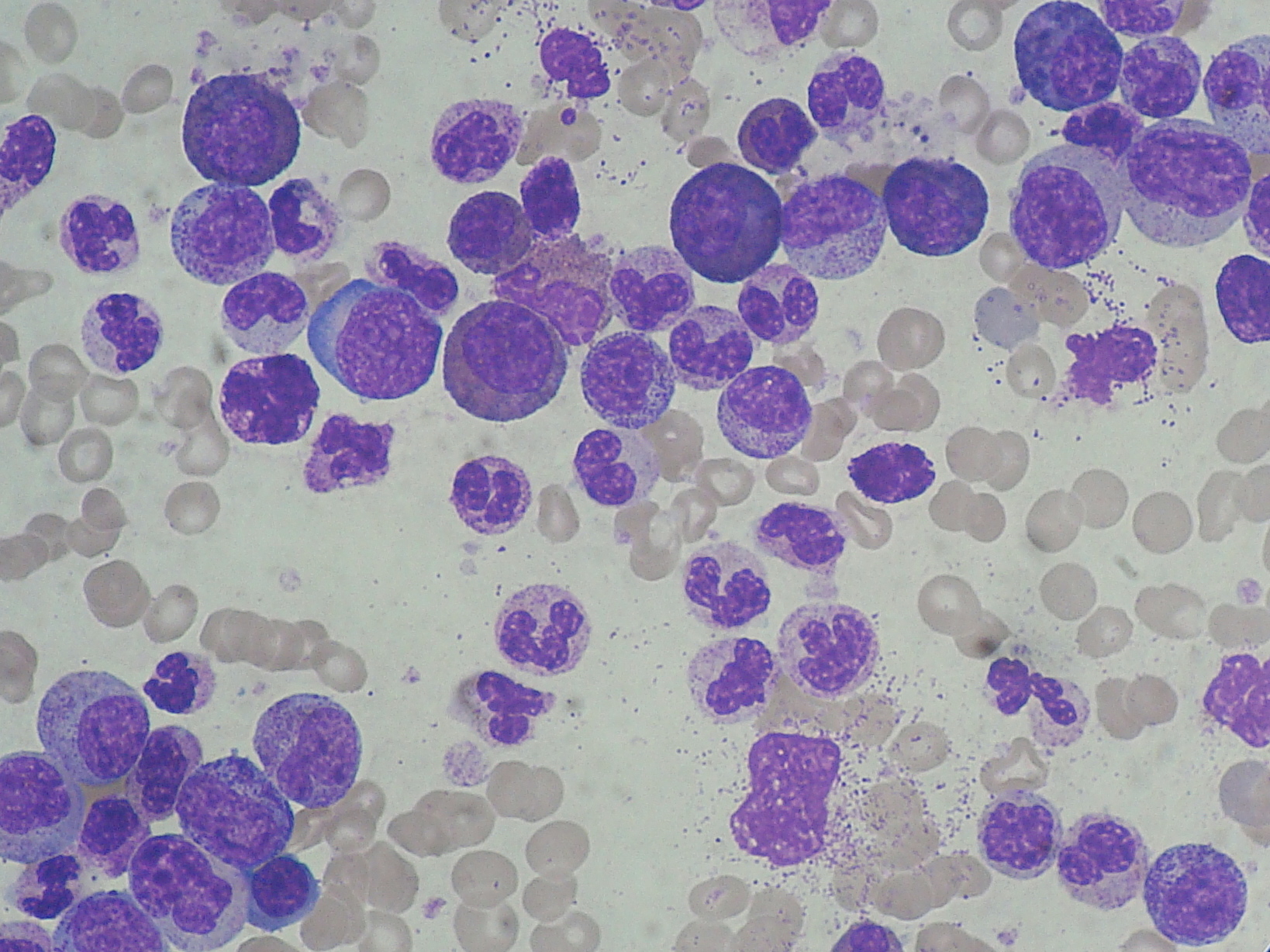{kind=link}
{kind=link}
{kind=link}
· Difference between chronic myelogenous leukemia (CML) and leukemoid reaction:
· a) Splenomegaly (a feature of CML; in the leukemoid reaction, it depends on the underlying disease)
· b) WBCs (usually > 100 000 /cmm on CML; usually < 50 000 /cmm on leukaemoid reaction)
· c) Myelocyte & neutrophil peaks (present on CML; absent on leukemoid reaction)
· d) Basophilia/eosinophilia/monocytosis (present on CML; absent on leukemoid reaction)
· e) Toxic granules (absent on CML; present on leukemoid reaction)
· f) LAP (Leukocyte alkaline phosphatase) score (low on CML; normal or increased on leukemoid reaction)
· g) Bone marrow examination (trilineage hyperplasia on CML; myeloid hyperplasia on leukemoid reaction)
· h) Genetic analysis (Philadelphia chromosome/ bcr – able fusion gene on CML; normal on leukemoid reaction).
· Polycythemia vera (PV, PCV; also known as erythremia, primary polycythemia, and polycythemia rubra vera): a neoplasm in which the bone marrow makes too many RBCs (red blood cells); it may also result in the overproduction of WBCs (white blood cells) and platelets. It is one of the myeloproliferative disorders.
· It occurs in all age groups, although the incidence increases with age. One study found the median age at diagnosis was 60 years, while a Mayo Clinic study found the highest incidence was among people aged 70–79 years. The overall incidence in Minnesota was 1.9 per 100,000 people/ year, and the disease was more common in men than women.
· Pathophysiology: polycythemia vera (PCV) is caused by neoplastic proliferation and maturation of erythroid, megakaryocytic, and granulocytic elements to produce panmyelosis. In contrast to secondary polycythemias, PCV is associated with low serum erythropoietin (EPO) levels. Instead, PCV cells have a mutation in the tyrosine kinase (JAK2), which acts in signaling pathways of the EPO receptor, rendering those cells hypersensitive to EPO.
· Calreticulin (CALR) mutations have been reported in Janus kinase 2 (JAK2) and myeloproliferative leukemia (MPL) – negative essential thrombocythemia and primary myelofibrosis. A study demonstrated that CALR mutations can also be associated with JAK2-negative PV (Reference: http://www.ncbi.nlm.nih.gov/pubmed/25305205 ).
· Signs & symptoms: patients with polycythemia vera may be asymptomatic. Pruritus (itching) occurs in about 40% of patients with PV, particularly after exposure to warm water (e.g., when taking a bath), and may result from abnormal histamine or prostaglandin release.
· Also, 20% may have gouty arthritis. Peptic ulcer disease (PUD) is also common. A rare but classic symptom of polycythemia vera (and also of essential thrombocythemia) is erythromelalgia, a sudden, severe burning pain in the hands or feet, usually accompanied by a reddish or bluish coloration of the skin caused by an increased platelet count or increased platelet "stickiness" (aggregation), resulting in the formation of tiny blood clots in the vessels of the extremity; it responds rapidly to treatment with aspirin.
· Patients with polycythemia vera are prone to the development of thrombosis and major thrombotic complications (e.g., myocardial infarction (MI, i.e., heart attack), stroke, deep vein thrombosis (DVT), pulmonary embolism (PE), or Budd–Chiari syndrome (hepatic vein thrombosis)] may sometimes be the first symptom or indication that a person has polycythemia vera.
· People with PV also have a risk of developing myelofibrosis.
· Headaches, lack of concentration, and fatigue are common symptoms.
· Diagnosis: a mutation in the JAK2 kinase (V617F) is strongly associated with polycythemia vera. JAK2 is a member of the Janus Kinase family and makes the erythroid precursors hypersensitive to erythropoietin (EPO).
· Polycythemia vera (PV) is associated most often with the JAK2V617F mutation in greater than 95% of cases, whereas the remainder has a JAK2 exon 12 mutation.
· Cellular and fibrotic phase.
· Other features that may be seen in PCV include a low ESR (erythrocyte sedimentation rate); and a raised leukocyte alkaline phosphatase (LAP). Note: LAP score is low on CML but normal or increased on leukemoid reaction.
· Also, the patient has elevated hemoglobin level and hematocrit (hematocrit may be as high as 70 to 80%), reflecting the increased number of red blood cells (there may be 8 to 9 million and occasionally 11 million erythrocytes per cubic millimeter of blood; a normal range for adults is 4 – 6), while the total blood volume sometimes increases as much as twice normal.
· The patient may also have an increased platelet count or WBCs (white blood cells). Because polycythemia vera results from an essential increase in erythrocyte production, patients have a low erythropoietin (EPO) level.
· Treatment: phlebotomy, often combined with other therapies. Removing blood from the body reduces blood volume and lowers hematocrit, reducing the risk of blood clots. Venesection is performed in people with polycythemia vera to bring their hematocrit (red blood cell percentage) down below 45 for men or 42 for women. It also improves cognitive impairment. Low-dose aspirin is often prescribed, as it reduces the risk of various thrombotic complications.
· Chemotherapy, usually with a "cytoreductive agent" such as hydroxycarbamide (hydroxyurea) may also be used.
· Erlotinib, an EGFR inhibitor, may also help, while selective JAK2 inhibitors such as ruxolitinib (Jakafi™, Jakavi™) are also being studied. Some medical practitioners tend to avoid chemotherapy, especially in young patients, as there is a possible increased risk of transformation to acute myeloid leukemia (AML).
· Note: Polycythemia: The World Health Organization (WHO) classification of hematologic malignancies defines polycythemia as Hb greater than 18.5 g/dL in men, greater than 16.5 g/dL in women, hematocrit greater than 99th percentile of the method-specific reference range, or Hb greater than 17 g/dL in men, greater than 15 g/dL in women with a documented and sustained increase of at least 2 g/dL from baseline not attributed to correction of iron deficiency.
· Absolute polycythemia is an increase in the total RBC mass in the body. Causes include hypoxia, inappropriate erythropoietin production, genetic polycythemia, and polycythemia vera.
· Relative polycythemia is an increase in hematocrit (Hct) or RBC count resulting from a decrease in plasma volume; however, total RBC mass is not increased. Causes include dehydration, shock, diuretic therapy, and spurious polycythemia (Gaisbock syndrome). Note: Gaisbock syndrome is a specific type of relative polycythemia primarily occurring in obese men in whom hypertension causes a reduction in plasma volume, resulting in (amongst other changes) a relative increase in red blood cell count.
· Causes of secondary polycythemia: the main reasons for higher-than-normal erythropoietin (EPO) are chronic hypoxia, poor oxygen delivery due to abnormal red blood cell structure, and tumors releasing excessive amounts of EPO inappropriately.
· Some of the common conditions that can result in elevated erythropoietin due to chronic hypoxia or poor oxygen supply include COPD (chronic obstructive pulmonary disease), hypoventilation syndrome, pulmonary hypertension, congestive heart failure (CHF), obstructive sleep apnea, kidney ischemia (e.g., renal artery stenosis), kidney transplantation, living in high altitudes, etc.
· Hemoglobinopathies with higher oxygen affinity by hemoglobin are another cause and include 2,3-BPG deficiency.
· Also, certain tumors have a tendency to secrete inappropriately high amounts of EPO, leading to polycythemia. The common Epo-releasing tumors are renal cell carcinoma, hepatocellular carcinoma, adrenal adenoma or carcinoma, and uterine cancer. More benign conditions may also cause increased EPO secretion, such as kidney cysts and kidney obstruction.
· Also, chronic carbon monoxide (CO) exposure, including long–term tobacco smoking, can lead to polycythemia. Hemoglobin naturally has a higher affinity for carbon monoxide than for oxygen.
· Polycythemia in newborns (neonatal polycythemia) is often caused by the transfer of maternal blood from the placenta or blood transfusions. Also, intrauterine hypoxia due to insufficiency of the placenta can lead to neonatal polycythemia (Reference (Retrieved December 2, 2015): http://www.emedicinehealth.com/polycythemia_high_red_blood_cell_count/page3_em.htm ).
· Essential thrombocythaemia (ET; also known as essential thrombocytosis, essential thrombocythemia, primary thrombocytosis): a rare chronic blood disorder characterized by the overproduction of platelets by megakaryocytes in the bone marrow. It is one of the myeloproliferative disorders. It may rarely develop into myelofibrosis or acute myeloid leukemia (AML). The incidence of ET is 0.6 – 2.5/100 000 per year; the median age at onset is 65 – 70 years and is more frequent in females than males. The incidence in children is 0.09/100 000 per year.
· Signs & symptoms: most people with ET are asymptomatic as the condition is usually discovered upon routine complete blood count (CBC). The most common symptoms are bleeding, thrombosis, leukocytosis, anemia, headache, nausea, vomiting, abdominal pain, visual disturbances, dizziness, fainting, splenomegaly, and numbness in the extremities.
· Pathophysiology: in ET, developing megakaryocytes are more sensitive to growth factors. Platelets derived from abnormal megakaryocytes are activated, which, along with the elevated platelet count, increases the likelihood of thrombosis. The increased risk of bleeding when the platelet count exceeds 1 million is due to von Willebrand factor (vWF) sequestration by the increased platelet mass, leaving insufficient vWF for platelet adhesion.
· Diagnosis: a mutation in the JAK2 kinase (V617F; member of the Janus kinase family) is present in 40 – 50% of cases.
· Essential thrombocythemia (ET) is associated with the JAK2V617F mutation in up to 55% of cases and with an MPL (thrombopoietin receptor) mutation in up to 5% of cases. Calreticulin (CALR) mutations have been reported in Janus kinase 2 (JAK2) and myeloproliferative leukemia (MPL) – negative essential thrombocythemia and primary myelofibrosis (Reference: http://www.ncbi.nlm.nih.gov/pubmed/25305205 ).
· Cellular phase and fibrotic phase.
· Platelet count is > 450 × 103/µL for at least 2 months.
· There is no cause for reactive thrombocytosis, and inflammatory indices are normal.
· There is also no evidence of iron deficiency; no evidence of polycythemia vera and no evidence of chronic myeloid leukemia (CML); however, the Philadelphia chromosome may be present in up to 10% of cases.
· Patients with the Philadelphia chromosome have a potential for the development of acute leukemia, especially acute lymphocytic leukemia (ALL).
· Less commonly than polycythemia vera, essential thrombocytopenia may lead to myelofibrosis.
· Treatment: Not all those affected will require aggressive treatment at presentation. Patients are usually split into low- and high-risk groups for bleeding/blood clotting (based on age, medical history, blood counts, and lifestyle). Low-risk individuals are usually treated with aspirin, whereas those at high risk are given hydroxycarbamide (hydroxyurea) and/or other treatments that reduce platelet counts, such as anagrelide and interferon-alpha.
· Primary myelofibrosis (PMF; also known as chronic idiopathic myelofibrosis and (primary) osteomyelofibrosis); a rare bone marrow cancer classified as a myeloproliferative neoplasm in which the proliferation of an abnormal clone of hematopoietic stem cells in the bone marrow and other sites results in fibrosis, i.e., the replacement of the marrow with collagenous connective tissue fibers.
· Primary myelofibrosis is the idiopathic form of the disease, in contrast with myelofibrosis secondary to polycythemia vera or (less commonly) essential thrombocytopenia. In some cases, It may be accelerated by the oral chemotherapy drug hydroxyurea. Myelofibrosis is a form of myeloid metaplasia. Agnogenic myeloid metaplasia and myelofibrosis with myeloid metaplasia (MMM) also overlap on the same spectrum.
· The disorder usually develops slowly and is mainly observed in people over the age of 50. It may also develop as a side effect of treatment with some drugs for hematological disorders.
· Signs & symptoms: abdominal fullness related to splenomegaly; bone pain; bruising and bleeding due to thrombocytopenia; fatigue; increased susceptibility to infections; and pallor and shortness of breath (due to anemia).
· Because of a high rate of cell turnover, hyperuricemia & gout may also occur. In rarer cases, MCV is elevated.
· Cutaneous myelofibrosis is a rare skin condition characterized by dermal and subcutaneous nodules.
· Lab tests/ CBC/ peripheral blood smear/ bone marrow: Primary myelofibrosis can begin with a blood picture suggestive of polycythemia vera (PV) or chronic myelogenous leukemia (CML). Most patients have moderate-to-severe anemia and eventually develop thrombocytopenia.
· The peripheral smear appears markedly abnormal, with the presentation of pancytopenia and teardrop-shaped RBCs, while nucleated erythroid precursors may be seen. Immature white cells are also seen, and basophils are increased.
· Bone marrow aspiration results in a dry tap, and there is reticulin or collagen fibrosis in the bone marrow biopsy.
· Genetics: there is an association between mutations in the JAK2, CALR, or MPL gene and myelofibrosis. Approximately 90% of those with myelofibrosis carry one of these mutations, and 10% carry none. These mutations are not specific to myelofibrosis and are linked to other myeloproliferative disorders, specifically essential thrombocythemia. The V617F mutation in the JAK2 protein is found in approximately 50% of individuals with primary myelofibrosis. The MPL gene encodes a protein that functions as a thrombopoietin receptor. A mutation in that gene, known as W515, produces an abnormal thrombopoietin receptor protein, resulting in the overproduction of abnormal megakaryocytes. Abnormal megakaryocytes stimulate other cells, such as fibroblasts, to produce collagen in the bone marrow.
· Calreticulin (CALR) mutations have been reported in Janus kinase 2 (JAK2) and myeloproliferative leukemia (MPL) – negative essential thrombocythemia and primary myelofibrosis (Reference: http://www.ncbi.nlm.nih.gov/pubmed/25305205 ).
· Pathogenesis: myelofibrosis is a clonal neoplastic disorder of hematopoiesis and is one of the myeloproliferative disorders. Production of cytokines (e.g., fibroblast growth factor (FGF)) by the abnormal hematopoietic cell clone (particularly by megakaryocytes) leads to the replacement of the hematopoietic tissue of the bone marrow by collagen fibrosis, impairing the patient's ability to generate new blood cells and resulting in progressive pancytopenia.
· Also, we have extramedullary hematopoiesis, i.e., blood cell formation occurring in sites other than the bone marrow, especially the liver (hepatomegaly) and spleen (splenomegaly). The last also contributes to causing pancytopenia, particularly thrombocytopenia and anemia. Another complication of extramedullary hematopoiesis is poikilocytosis.
· Treatment: the one known curative treatment is allogeneic stem cell transplantation. Other supportive treatment options include dexamethasone, interferon-alpha, hydroxycarbamide (hydroxyurea), thalidomide, and lenalidomide (the last 2 may cause peripheral neuropathy).
· Splenectomy is sometimes considered a treatment option for patients with myelofibrosis in whom massive splenomegaly contributes to anemia; however, splenectomy – in the presence of massive splenomegaly – is a high-risk procedure, with a mortality risk as high as 3% in some studies. Ruxolitinib (Jakafi™, Jakavi™), an inhibitor of JAK1 and JAK2, represents a new therapeutic approach.
· Mastocytosis: one of the mast cell diseases; a rare disorder of children and adults caused by too many mast cells and CD34+ mast cell precursors.
· People affected by mastocytosis are susceptible to itching, hives, and anaphylactic shock, caused by the release of histamine from mast cells. Mastocytosis can occur in a variety of forms. Most cases are cutaneous (confined to the skin only), and there are several forms. The most common cutaneous mastocytosis is urticaria pigmentosa (UP), which is more common in children.
· Telangiectasia macularis eruptiva perstans (TMEP) is a much rarer form of cutaneous mastocytosis that affects adults.
· UP and TMEP can evolve into indolent systemic mastocytosis. Systemic mastocytosis involves the bone marrow in some cases and other internal organs in others, usually in addition to the skin. Any organ can be involved. Mast cells accumulate in various tissues and can affect organs such as the liver, spleen, and lymph nodes, as well as enlarged organs. In the bowel, it may manifest as mastocytic enterocolitis.
· Signs & symptoms: The symptoms of mastocytosis resemble those of the symptoms of an allergic reaction and may include fatigue, urticaria pigmentosa, itching, abdominal discomfort, diarrhea, food and blood intolerance, infections (bronchitis, rhinitis, and conjunctivitis), ear/nose/throat inflammation, anaphylaxis, hypotension and faintness, bone or muscle pain, osteoporosis or osteosclerosis, malabsorption, ocular discomfort, headache, etc.
· Diagnosis: by taking a biopsy from a different organ, such as the bone marrow (increase in mast cells).
PLASMA CELL DYSCRASIAS
· Plasma cell dyscrasias: disorders of the plasma cells produced as a result of abnormal proliferation of a monoclonal population of plasma cells that may or may not secrete detectable levels of a monoclonal immunoglobulin or immunoglobulin fragment (paraprotein or M protein). The most common plasma cell dyscrasia is monoclonal gammopathy of undetermined significance (MGUS).
· Classification:
· Cryoglobulinemia
· Heavy chain disease
· Monoclonal gammopathy of undetermined significance (MGUS)
· Multiple myeloma
· Plasmacytoma
· Plasma cell leukemia
· POEMS syndrome
· Primary amyloidosis (immunoglobulin light chain amyloidosis)
· Waldenstrom’s macroglobulinemia.
· Note: POEMS syndrome, also known as Crow–Fukase syndrome, Takatsuki disease, or PEP syndrome, is a rare syndrome defined as the combination of a plasma-cell proliferative disorder, typically myeloma (usually osteosclerotic, contrary to osteolytic bone lesions in other myelomas), polyneuropathy and effects on many other organ systems. The name is an acronym deriving from some of the main features: Polyneuropathy (peripheral nerve damage), Organomegaly, Endocrinopathy or Edema, M-protein (an abnormal immunoglobulin), and Skin abnormalities. However, these features are neither an exhaustive list nor seen in all individuals with the condition.
· Cryoglobulinemia (CG): a medical condition in which the blood contains large amounts of cryoglobulins – proteins (mostly immunoglobulins) that become insoluble at reduced temperatures. They usually consist of IgM directed against the Fc region of IgG. This should be contrasted with cold agglutinins (see above), which cause RBC agglutination. Cryoglobulins typically precipitate at temperatures below normal body temperature (37°C) and will dissolve again if the blood is heated.
· Cryoglobulinemia can be associated with various diseases such as multiple myeloma and hepatitis.
· Cryoglobulinemia is part of a group of diseases that cause damage and inflammation of the blood vessels throughout the body (vasculitis). There are three main types of cryoglobulinemia. They are grouped based on the type of antibody that is produced (Brouet classification):
· Type I (10 – 15% of cases)
· Type II (50 – 60% of cases)
· Type III (25 – 30% of cases)
· Types II and III are also referred to as mixed cryoglobulinemia.
{kind=link}
· For classification, see also:
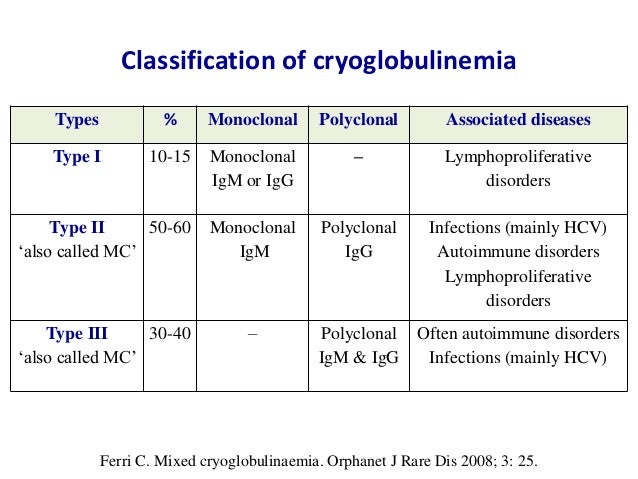{kind=link}
· Type I cryoglobulinemia is most often related to cancer of the blood or immune systems. Type I cryoglobulins are composed of isolated monoclonal immunoglobulins. It is most commonly encountered in patients with plasma cell dyskrasia such as multiple myeloma or Waldenstrom’s macroglobulinemia.
· Type II cryoglobulins are composed of immunocomplexes formed by monoclonal IgM.
· Type III - cryoglobulins are composed of immunocomplexes formed by polyclonal IgM.
· Types II and III are most often found in people with chronic inflammatory conditions, such as autoimmune disease or hepatitis C. Most people with this form of the disease have a chronic hepatitis C infection.
· Type III is strongly associated with autoimmune diseases such as SLE (systemic lupus erythematosus) and RA (rheumatoid arthritis).
· Cryoglobulins may be present in Mycoplasma pneumonia, post-streptococcal glomerulonephritis, multiple myeloma, certain leukemias, primary macroglobulinemia, and some autoimmune diseases such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE), and as a symptom in 35% of chronic hepatitis C infections; they may also occur in hepatitis B and HIV infections.
· Hyperviscosity is typically associated with CG due to hematological malignancies and monoclonal immunoglobulins.
· Meltzer’s triad (purpura, arthralgia, weakness) is generally seen with polyclonal CGs and is seen in essential, viral, or connective tissue disease-associated CG.
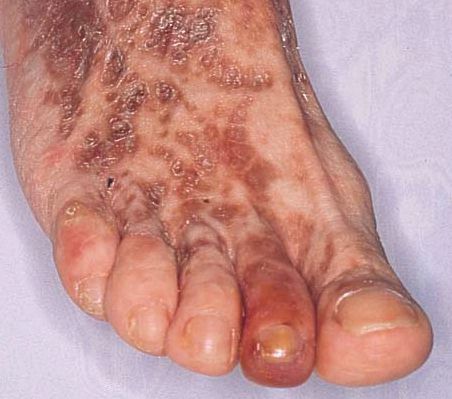
· Symptoms: symptoms will vary depending on the disorder type and the organs involved. Symptoms may include dyspnea, fatigue, glomerulonephritis, joint & muscle pain, purpura, Raynaud’s phenomenon, skin necrosis, and skin ulcers.
· Lab τests: CBC (FBC), complement (numbers will be low), cryoglobulin test (may show the presence of cryoglobulins), liver function tests (may be high if hepatitis C is present), RF (positive in types II and III), a skin biopsy (may show vasculitis), protein electrophoresis (may show an abnormal antibody protein), urinalysis (may show hematuria if the kidneys are affected), HCV (hepatitis C) serology, ESR.
· Other tests may include an angiogram, CXR (chest x-ray), and nerve conduction tests if the person has weakness in the arms or legs.
· Complications include bleeding in the digestive tract (rare), heart disease (rare), infections of ulcers, kidney failure, liver failure, skin death, and death.
· Differential diagnosis with cold agglutinin disease: Cryoglobulinemia has almost none of the features of cold agglutinin disease, except for a history of Raynaud syndrome and an elevated IgM level in some cases (but without hemolysis).
· Treatment:
· Mixed cryoglobulinemia (Types II and III): mild or moderate forms of cryoglobulinemia can often be treated by taking steps to deal with the underlying cause. Mild cases can be treated by avoiding cold temperatures. Standard hepatitis treatments help most people with hepatitis C who have mild or moderate disease. The condition may relapse when treatment stops. Severe cryoglobulinemia involves vital organs or large areas of skin. It is treated with corticosteroids and other medicines that suppress the immune system. Rituximab is an effective drug and has fewer risks than other medicines. Cyclophosphamide is used for life-threatening conditions when rituximab is ineffective or unavailable. This medicine was used often in the past. Also, plasmapheresis may be used. In this procedure, blood plasma is removed from circulation, and abnormal cryoglobulin antibodies are removed. Plasma is replaced by fluid, protein, or donated plasma.
· Type I cryoglobulinemia: this disorder is due to cancer of the blood or immune system, such as multiple myeloma. Treatment targets the abnormal cancer cells that produce cryoglobulin.
· Prognosis: most of the time, mixed cryoglobulinemia does not lead to death. The prognosis can be poor if the kidneys are affected.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Multiple myeloma (MM): a neoplasm of plasma cells characterized by the formation of multifocal tumor masses composed of plasma cells at multiple (bone marrow–based) locations in the skeleton; suppression of normal immunoglobulin production (hypogammaglobulinemia); and production of a monoclonal protein (paraprotein; M protein).
· Multiple myeloma develops in 6.1 per 100,000 people per year; it is more common in men and, for unknown reasons, is twice as common in African Americans as in Caucasians.
· The average age of onset for multiple myeloma is 70 years.
· Clinical features: age 50 – 60 years; back pain, spontaneous fractures, renal failure, anemia, infections, bleeding; hyperviscosity syndrome.
· A mnemonic for some common symptoms of multiple myeloma is CRAB: C = Calcium (elevated), R = Renal failure, A = Anemia, B = Bone lesions. Myeloma can cause many other symptoms, including opportunistic infections. Bone pain affects almost 70% of patients and is the most common symptom, usually involving the spine and ribs. Persistent localized pain may indicate a pathological bone fracture, while the involvement of the vertebrae may lead to spinal cord compression. Plain radiographs may show "punched-out" resorptive lesions, including the "pepper pot" appearance of the skull. The breakdown of bone also releases calcium into the blood, leading to hypercalcemia.
· Anemia is usually normocytic & normochromic.
· The patient may develop acute or chronic kidney failure. Increased bone resorption leads to hypercalcemia and causes nephrocalcinosis, contributing to kidney failure. Amyloidosis may also contribute to kidney injury.
· Light chains produce many effects, which can manifest as Fanconi syndrome (type II renal tubular acidosis). Other causes of kidney injury include hyperuricemia, pyelonephritis, and local infiltration of tumor cells.
· Headache, visual changes, and retinopathy may be the result of hyperviscosity of the blood, depending on the properties of the paraprotein.
· Lab features/ CBC/ Peripheral blood smear:
· Blood smear: Rouleaux formation.
· Bone marrow: plasma cells> 10%.
· Other lab features: elevated ESR.
· A high level of CRP is often seen in myeloma patients and increases with active disease.
· Uric acid may also be elevated.
· Increased BUN & creatinine show renal disease.
· Serum/urine protein electrophoresis: M band. In serum protein electrophoresis, separation of different proteins is achieved based on their charge. The serum is applied to the cellulose acetate membrane, which is then placed in an electrophoresis apparatus. Normally, 5 zones of proteins can be distinguished from the anode (–) to the cathode (+): albumin, alpha1 – globulin, alpha2 – globulin, beta – globulin, and gamma – globulin. Immunoglobulins migrate to the gamma region. In multiple myeloma, electrophoresis shows an M-band and a spike. A localized dense band with sharp margins in the gamma (γ) region indicates the M band.
· One type of paraprotein is the Bence Jones protein, a urinary paraprotein composed of free light chains.
· Serum free light chains (FLC) is a test that measures the amount of free light chains in the blood. Most people with multiple myeloma produce increased amounts of either kappa or lambda-free light chains, which can be measured in blood. Consequently, the ratio of kappa to lambda light chains is abnormal in most people and is a sensitive indicator for this disease (Reference (Retrieved: December 2, 2015): https://labtestsonline.org/understanding/conditions/mult-myeloma/start/2 )
· On the densitometric scan, a narrow–based, tall, sharply defined spike is seen.
· X-Ray: localized osteolytic lesion.
· Diagnostic criteria: updated criteria of the International Myeloma Working group for symptomatic myeloma, asymptomatic myeloma, and MGUS (monoclonal gammopathy of undetermined significance):
· I) Symptomatic myeloma:
· i) Clonal plasma cells >10% on bone marrow biopsy or (in any quantity) in a biopsy from other tissues (plasmacytoma).
· ii) A monoclonal protein (paraprotein) in either serum or urine (except in cases of true non-secretory myeloma).
· iii) Evidence of end-organ damage felt related to the plasma cell disorder: related organ or tissue impairment, ROTI, commonly referred to by the acronym "CRAB": hyperCalcaemia (corrected calcium > 2.75 mmol/L); Renal insufficiency related to myeloma; Anemia (hemoglobin <10 g/dL); Bone lesions (lytic lesions or osteoporosis with compression fractures).
· Note: recurrent infections alone in a patient who has none of the CRAB features are not sufficient to make the diagnosis of myeloma. Patients who lack CRAB features but have evidence of amyloidosis should be considered amyloidosis, not myeloma.
· Characteristics of multiple myeloma:
· a) M protein appears as a narrow spike in the gamma, beta, or alpha2 regions.
· b) M-protein level is usually greater than 3 g/ dL.
· c) Skeletal lesions (e.g., lytic lesions, diffuse osteopenia, vertebral compression fractures) are present in 80% of patients.
· d) Diagnosis requires 10 – 15% plasma cell involvement on bone marrow biopsy.
· e) Anemia, pancytopenia, hypercalcemia, and renal disease may be present.
· II) Asymptomatic / smoldering myeloma:
· 1) Serum paraprotein >30 g/L (> 3g/dL) and/or
· 2) Clonal plasma cells >10% on bone marrow biopsy and
· 3) NO myeloma-related organ or tissue impairment. Affected patients have no lytic bone lesions, anemia, hypercalcemia, or renal disease.
· ΙΙΙ) Monoclonal gammopathy of undetermined significance (MGUS):
· i) Serum paraprotein <30 g/L (< 3g/dL) AND
· ii) Clonal plasma cells <10% on bone marrow biopsy AND
· iii) NO myeloma-related organ or tissue impairment. Related conditions include solitary plasmacytoma (a single tumor of plasma cells, typically treated with radiotherapy), plasma cell dyscrasia (where only the antibodies produce symptoms, e.g., amyloid light chain (AL) amyloidosis), and POEMS syndrome (peripheral neuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, skin changes).
· IV) Heavy chain disease
· The M protein has an incomplete heavy chain and no light chain.
· V) Plasma cell leukemia
· Peripheral blood contains more than 20% plasma cells.
· M-protein levels are low
· Affected patients have few bone lesions and few hematologic disturbances.
· This monoclonal gammopathy occurs in younger patients.
· VI) Solitary plasmacytoma
· Affected patients have only one tumor, with no other bone lesions and no urine or serum abnormalities.
· Staging: International Staging System (International Myeloma Working Group):
· Stage I: beta – 2 microglobulin (β2M) < 3.5 mg/L, albumin ≥ 3.5 g/dL
· Stage II: β2M < 3.5 mg/L and albumin < 3.5 g/dL or
· β2M (beta 2 microglobulin) 3.5–5.5 mg/L irrespective of the serum albumin
· Stage III: β2M ≥ 5.5 mg/L.
· Note that the ISS should be used only in patients who meet diagnostic criteria for myeloma. Patients with MGUS and asymptomatic myeloma who have kidney dysfunction from unrelated causes, such as diabetes or hypertension, may have elevated β2M levels from the kidney dysfunction and cannot be considered as stage III myeloma.
· It is recommended that the ISS be used along with the Durie-Salmon Staging System.
· Staging according to the Durie-Salmon Staging System:
· Stage I: all of: Hb > 10g/dL; normal calcium; skeletal survey: normal or single plasmacytoma or osteoporosis; serum paraprotein level < 5 g/dL if IgG, < 3 g/dL if IgA; and urinary light chain excretion < 4 g/24h.
· Stage II: fulfilling the criteria of neither I nor III], stage III [one or more of Hb < 8.5g/dL; high calcium > 12 mg/dL; skeletal survey: Three or more lytic bone lesions; serum paraprotein > 7g/dL if IgG, > 5 g/dL if IgA; and urinary light chain excretion > 12g/24h.
· Stages I, II, and III of the Durie-Salmon staging system can be divided into A or B depending on serum creatinine:
· A: serum creatinine < 2 mg/dL (< 177 μmol/L)
· B: serum creatinine > 2 mg/dL (> 177 μmol/L)
· Treatment: remissions may be induced with chemotherapy, steroids, proteasome inhibitors, immunomodulatory drugs (such as thalidomide & lenalidomide), and stem cell transplantation.
· Radiotherapy is sometimes used to reduce pain from bone lesions. Also, bisphosphonates (e.g., pamidronate or zoledronic acid) are routinely administered to prevent fractures; they have also been observed to have a direct anti-tumor effect even in patients without known skeletal disease.
· High-dose chemotherapy with autologous hematopoietic stem–cell transplantation (ASCT) has become the preferred treatment for patients under the age of 65. Today's most common induction regimens are lenalidomide–dexamethasone and bortezomib-based regimens. In younger patients, post-ASCT maintenance therapy with thalidomide appears to increase tumor burden reduction further, which translates into prolonged PFS (progression-free survival).
· Patients over age 65 and patients with a significant concurrent illness often cannot tolerate stem cell transplantation, so the standard of care for these patients has been chemotherapy with melphalan and prednisone. However, improved outcomes may also occur with new chemotherapy regimens, e.g., with bortezomib.
· For patients with relapsed disease, options include bortezomib (a proteasome inhibitor), lenalidomide (|a less toxic thalidomide analog), or the newly approved thalidomide derivative pomalidomide.
· Prognosis: With high-dose therapy followed by autologous hematopoietic stem–cell transplantation (ASCT) the median survival is about 4.5 years, compared to a median of approximately 3.5 years with "standard" therapy.
· Overall, the 5-year survival rate is around 35%. Advanced treatments may extend median survival to 5 – 7 years or longer. The average age of onset for multiple myeloma is 70 years.
· Older patients often experience other serious diseases, which affect survival. Younger patients might have much longer survival rates.
· Monoclonal gammopathy of undetermined significance (MGUS; formerly called benign monoclonal gammopathy): a condition in which a paraprotein is found in the blood during standard laboratory tests. It resembles multiple myeloma and similar diseases, but antibody levels are lower, the number of plasma cells (differentiated B cells that secrete antibodies) in the bone marrow is lower, it has no symptoms or problems, and no treatment is indicated.
· However, multiple myeloma develops at the rate of about 1 – 2% a year, so doctors recommend monitoring it yearly. It may also be related to a slowly progressive symmetric distal sensorimotor neuropathy in rare cases.
· MGUS is a common, age-related medical condition characterized by an accumulation of bone marrow plasma cells derived from a single abnormal clone.
· Diagnostic criteria: Patients may be diagnosed with MGUS if they fulfill the following four criteria:
· i) A monoclonal paraprotein band less than 30 g/L (< 3g/dL).
· ii) Plasma cells < 10% on bone marrow examination.
· ii) No M protein in their urine; no evidence of (lytic) bone lesions, anemia, hypercalcemia, or renal insufficiency related to the paraprotein.
· iv) No evidence of another B-cell proliferative disorder.
· Pathology: Pathologically, the lesion in MGUS is, in fact, similar to that in multiple myeloma. There is a predominance of clonal plasma cells in the bone marrow with an abnormal immunophenotype (CD38+ CD56+CD19–) mixed in with cells of a normal phenotype (CD38+ CD56− CD19+). In MGUS, on average, >3% of clonal plasma cells have the normal phenotype, whereas in multiple myeloma, <3% have the normal phenotype.
· Prognosis: a study of the Mayo Clinic showed that MGUS transformed into multiple myeloma or a similar lymphoproliferative disorder at the rate of about 1 – 2% a year, or 17%, 34%, and 39% at 10, 20, and 25 years, respectively, of follow-up—among surviving patients. Only 11.2% of the patients developed lymphoproliferative disorders, and most of the patients (most of them were elderly) died from other causes. In 2009, prospective data demonstrated that all or almost all cases of multiple myeloma are preceded by MGUS.
· In addition to multiple myeloma, MGUS may also progress to Waldenstrom’s macroglobulinemia, primary amyloidosis, B–cell lymphoma, or chronic lymphocytic leukemia (CLL).
· Management: the protein electrophoresis test should be repeated annually, and if there is any concern for a rise in monoclonal protein level, then prompt referral to a hematologist is indicated. When first evaluating a case of MGUS, the hematologist will usually perform a skeletal survey (X-rays of the proximal skeleton), check the blood for hypercalcemia and deterioration of renal function, check the urine for Bence Jones protein, and perform a bone marrow biopsy. If none of these tests are abnormal, a patient with MGUS is followed up once every 6 months to a year with serum protein electrophoresis.
· Waldenstrom's macroglobulinemia (WM, also known as lymphoplasmacytic lymphoma): cancer affecting B cells. The main attributing antibody is immunoglobulin M (IgM). WM is an "indolent lymphoma" (i.e., it tends to grow and spread slowly).
· WM is a rare disease, with an incidence rate of about 3 cases per million people per year in the United States, with only about 1,500 cases per year in the U.S. WM is a type of lymphoproliferative disease, which shares clinical characteristics with the indolent non – Hodgkin’s lymphoma.
· As with other lymphomas, the disease is characterized by an uncontrolled increase of B cells in the bone marrow and lymph nodes. The proliferation of B-cells interferes with RBC production, resulting in anemia. A unique characteristic of the disease is that B-cells produce excess amounts of the immunoglobulin protein (IgM), which causes hyperviscosity (thickening of the blood) and requires additional treatment.
· Diagnosis: blood tests show the level of IgM in the blood and the presence of proteins, or tumor markers, which are the key symptoms of WM.
· Bone marrow biopsy: shows particular lymphocytes that indicate WM.
· Flow cytometry may be used to examine markers on the cell surface or inside the lymphocytes.
· Lab features: The ESR and uric acid levels may be elevated. Creatinine is occasionally elevated, and approximately 4% of patients have high blood calcium. The LDH level is frequently elevated.
· Beta - 2 – microglobulin and CRP are not specific for Waldenström macroglobulinemia. Beta-2-microglobulin is elevated in proportion to the tumor mass.
· Serum protein electrophoresis results indicate evidence of a monoclonal spike but cannot establish the spike as IgM. An M component with beta-to-gamma mobility is highly suggestive of WM.
· Immunoelectrophoresis and immunofixation studies help identify the type of immunoglobulin, the light chain's clonality, and the paraprotein's monoclonality and quantitation.
· High-resolution electrophoresis and serum and urine immunofixation are recommended to help identify and characterize the monoclonal IgM paraprotein. The light chain of the monoclonal protein is usually the kappa light chain. At times, patients with WM may exhibit more than one M protein.
· Characterization studies of urinary immunoglobulins indicate that light chains (Bence Jones protein), usually of the kappa type, are found in the urine.
· Bence Jones proteinuria is observed in approximately 40% of patients and exceeds 1 g/d in approximately 3% of patients.
· Criteria for diagnosis of Waldenstrom macroglobulinemia:
· i) IgM monoclonal gammopathy that excludes chronic lymphocytic leukemia (CLL) and mantle cell lymphoma.
· ii) Evidence of anemia, constitutional symptoms, hyperviscosity, lymphadenopathy, or hepatomegaly/ splenomegaly that can be attributed to an underlying lymphoproliferative disorder.
· Hypercellular bone marrow with extensive infiltration by lymphoma cells.
· Prognosis: While the disease is incurable, it is treatable. Because of its indolent nature, many patients can lead active lives and, when treatment is required, may experience years of symptom-free remission. Current medical treatments result in survival for some longer than 10 years, partly because improved diagnostic testing enables earlier diagnosis and treatment.
LYMPHOMAS
· Lymphomas: a group of blood cell tumors that develop from lymphatic tissues.
· Symptoms may include enlarged lymph nodes (lymphadenopathy) that are not generally painful, fever, sweats (most common at night), pruritus (itchiness), weight loss, and fatigue.
· There are dozens of subtypes of lymphomas. The two main categories of lymphomas are Hodgkin lymphomas (HL) and non–Hodgkin lymphomas (NHL). WHO also includes multiple myeloma and other lymphoproliferative diseases (LPDs) (see above).
· The many subtypes of non-Hodgkin lymphomas make up about 90% of all lymphoma cases.
· Signs & symptoms include lymphadenopathy (swelling of lymph nodes; the primary presentation in lymphoma), and B – symptoms (systemic symptoms) that can be associated with both Hodgkin lymphoma and non-Hodgkin lymphoma. They consist of fever, night sweats, and weight loss. Other symptoms include anorexia, fatigue, and dyspnea.
· Diagnosis: Lymphoma is definitively diagnosed by an LN (lymph node) biopsy where a partial or total excision of an LN is examined under the microscope, and the examination reveals histopathological features that may indicate lymphoma. After lymphoma is diagnosed, a variety of tests may be carried out, including immunophenotyping, flow cytometry, and fluorescence in situ hybridization (FISH).
· Classification: classification systems classify lymphoma according to whether it is a Hodgkin lymphoma, whether the cell that is replicating is a T cell or B cell, and the site from which the cell arises.
· Subtypes:
· a) Mature B cell neoplasms
· b) Mature T cell & NK (natural killer) cell neoplasms
· c) Precursor lymphoid neoplasms
· d) Hodgkin’s lymphoma (HL)
· e) Immunodeficiency–associated lymphoproliferative disorders.
· Ann Arbor staging is the staging system for HL and NHL:
· i) Stage I: the cancer is located in a single region, usually one lymph node and the surrounding area; stage I often will not have outward symptoms
· ii) Stage II: indicates that the cancer is located in two separate regions, an affected lymph node or organ and a second affected area, and that both affected areas are confined to one side of the diaphragm - that is, both are above the diaphragm, or both are below the diaphragm
· iii) Stage III: cancer has spread to both sides of the diaphragm, including one organ or area near the lymph nodes or the spleen
· iv) Stage IV: diffuse or disseminated involvement of one or more extra lymphatic organs, including any involvement of the liver, bone marrow, or nodular involvement of the lungs.
· Modifiers (these letters can be appended to some stages):
· A or B: the absence of constitutional (B-type) symptoms is denoted by adding an "A" to the stage; the presence is denoted by adding a "B" to the stage
· S: if the disease has spread to the spleen
· E: if the disease is "extranodal" (not in the lymph nodes) or has spread from lymph nodes to adjacent tissue
· X: if the largest deposit is >10 cm large ("bulky disease"), or whether the mediastinum is wider than 1/3 of the chest on a CXR (chest X-ray)
· Treatment:
· I) HL (see).
· II) NHL (see).
· III) Low-grade lymphomas: many low-grade lymphomas remain indolent for many years. Treatment of the non-symptomatic patient is often avoided. Watchful waiting is often the initial course of action in these forms of lymphoma, such as follicular lymphoma. If a low-grade lymphoma is becoming symptomatic, radiotherapy or chemotherapy are the treatments of choice; although they do not cure the lymphoma, they can alleviate the symptoms. Some centers advocate using single-agent rituximab to treat follicular lymphoma rather than wait-and-watch. Watchful waiting is not a good strategy for all patients, as it has been equated with watch and worry in some patients.
· II) High-grade lymphomas: aggressive chemotherapy, including the CHOP or R – CHOP regimen. A number of people are cured with first-line chemotherapy. Most patients relapse within the first two years. For people who relapse, treatment includes high-dose chemotherapy followed by autologous stem cell transplantation.
· Prognosis according to stage at diagnosis:
· a) Localized (27% of cases): 82.1% five-year relative survival.
· b) Regional (spread to regional lymph nodes; 19% of cases): 77.5% five-year relative survival.
· c) Distal (metastasized 45% of cases): 59.9% five-year relative survival.
· d) Unknown (unstaged; 9% of cases): 67.5% five-year relative survival.
· Hodgkin’s lymphoma (HL): a type of lymphoma that originates from lymphocytes. A history of infectious mononucleosis due to infection by EBV (Epstein–Barr virus) may increase the risk of HL, but the precise contribution of EBV remains largely unknown.
· Approximately 0.2% of men and women will be diagnosed with Hodgkin lymphoma at some point during their lifetime, based on 2010 – 2012 data (Reference (Retrieved: December 4, 2015): http://seer.cancer.gov/statfacts/html/hodg.html ).
· It is characterized by the orderly spread of disease from one LN (lymph node) group to another and by the development of systemic symptoms with advanced disease.
· Signs & Symptoms: lymphadenopathy (enlargement of one or more lymph nodes; the lymph nodes may also feel rubbery and swollen when examined; cervical & supraclavicular nodes are most frequently involved; also lymph nodes of the chest are often affected and may be noticed on a CXR (chest – X Ray)), pruritus (itchiness), night sweats, weight loss, splenomegaly (30%, seldom massive)/ hepatomegaly (5%)/ hepatosplenomegaly, pain following alcohol consumption (2 – 3% of patients with HL; pathognomic sign of Hodgkin lymphoma; typically has an onset within minutes after ingesting alcohol, and is usually felt as coming from the vicinity where there is an involved lymph node; it been described as either sharp and stabbing or dull and aching), non-specific back pain, red-colored patches on the skin, easy bleeding and petechiae due to low platelet count; lassitude (fatigue), cyclical high-grade fever known as the Pel – Ebstein fever ("P-E fever"; however there is a debate about if it truly exists); and nephritic syndrome (most commonly: minimal change disease).
· Systemic symptoms such as fever, night sweats, and weight loss are known as B symptoms.
· LN biopsy: Microscopic examination of the lymph node (LN) biopsy reveals complete or partial effacement of the lymph node architecture by scattered large multinucleated malignant cells known as Reed – Sternberg (RCS) cells (typical and variants) that are large and are either multinucleated or have a bilobed nucleus with prominent eosinophilic inclusion-like nucleoli, resembling an "owl's eye" appearance.
{kind=link}
· Classification:
· a) Nodular sclerosing HL
· b) Mixed–cellularity subtype, lymphocyte–rich or lymphocytic predominance
· c) Lymphocyte depletes
· d) Unspecified.
· Note: nodular lymphocyte-predominant HL expresses CD20 and is not currently considered a form of classical Hodgkin's. For the other forms, although the traditional B cell markers such as CD20 are not expressed on all cells, Reed–Sternberg cells are usually of B cell origin.
· Staging: A patient will be staged after Hodgkin lymphoma is diagnosed. Staging may include documentation of histology, physical examination, blood tests, radiographs, CT/PET/MRI scans of the chest, abdomen, and pelvis, and usually a bone marrow biopsy.
· On the basis of this staging, the patient will be classified according to a staging classification, the Ann Arbor staging classification (see also above):
· Stage I: involvement of a single lymph node region (I) (mostly the cervical region) or single extra lymphatic site (Ie)
· Stage II: involvement of two or more lymph node regions on the same side of the diaphragm (II) or of one lymph node region and a contiguous extra lymphatic site (IIe)
· Stage III: involvement of lymph node regions on both sides of the diaphragm, which may include the spleen (IIIs) and/or limited contiguous extra lymphatic organ or site (IIIe, IIIes)
· Stage IV: disseminated involvement of one or more extra lymphatic organs.
· Treatment: Hodgkin lymphoma may be treated with chemotherapy, radiotherapy, or hematopoietic stem cell transplantation.
· Chemotherapy includes the classical MOPP [Mustargen (chlormethine), Oncovin (vincristine), Prednisone, Procarbazine]; the preferred scheme in the USA, ABVD (Adriamycin (doxorubicin), Bleomycin, Vinblastine, Dacarbazine); the preferred scheme in Europe, BEACOPP [Bleomycin, Etoposide, Adriamycin (doxorubicin), Cyclophosphamide, Oncovin (vincristine), Procarbazine and Prednisone]; and the Stanford V scheme [Doxorubicin, Vinblastine, Mechlorethamine, Vincristine, Bleomycin, Etoposide, Prednisone]. The common non-Hodgkin's lymphoma treatment, rituximab (a monoclonal antibody against CD20), is not routinely used to treat Hodgkin's lymphoma because most cases lack the CD20 surface antigen. However, the use of rituximab in Hodgkin's lymphoma, including the lymphocyte-predominant subtype, has recently been reviewed.
· Prognosis: the overall 5-year survival rate is 85%. There have been many cases of individuals living >40 years after diagnosis. In one recent European trial, the 5-year survival rate for patients with a favorable prognosis was 98%, while that for those with a worse outlook was at least 85%.
· The adverse prognostic factors identified in the international study are:
· a) Age ≥ 45 years
· b) Stage IV disease
· c) Hb<10.5 g/dl
· d) Lymphocytes < 600/μl or < 8%
· e) Male gender
· f) Albumin < 4 g/dl
· g) WBCs >_ 15 000/ μl.
· Non-Hodgkin lymphomas (NHLs): a diverse group of blood cancers that include any kind of lymphoma except Hodgkin’s lymphoma (HL). Types of NHL vary significantly in their severity, from slow-growing to very aggressive types. In the USA, data show that there were about 19.7 cases of NHL per 100,000 adults per year; and 6.3 deaths per 100,000 adults per year.
· About 2.1% of men and women will be diagnosed with NHL at some point during their lifetime, based on 2010 – 2012 data (Reference (Retrieved: December 4, 2015): http://seer.cancer.gov/statfacts/html/nhl.html ).
· Non-Hodgkin lymphoma increases with age steadily. Up to 45 years NHL is more common among males than females.
· Possible causes and associations of some forms of NHL include:
· a) Infectious agents: EBV [Epstein – Barr virus; associated with Burkitt’s lymphoma, Hodgkin’s lymphoma (HL), follicular dendritic cell sarcoma, extranodal NK – T – cell lymphoma], HTLV [Human T – cell lymphotropic virus) virus; associated with adult T–cell lymphoma], Helicobacter pylori (a bacterium well known as a causative microbe for peptic (stomach/ duodenum) ulcer disease, but also associated with gastric lymphoma), Kaposi's sarcoma-associated herpesvirus [KSHV; also known as HHV8 (Human Herpes Virus 8); associated with primary effusion lymphoma, multicentric Castleman disease], HCV (hepatitis C virus; associated with splenic marginal zone lymphoma, lymphoplasmacytic lymphoma and diffuse large B – cell lymphoma), and HIV.
· Note: Castleman disease (also known as giant or angiofollicular lymph node hyperplasia, lymphoid hamartoma, angiofollicular lymph node hyperplasia) is a group of uncommon lymphoproliferative disorders that share common lymph node histological features that may be localized to a single lymph node (unicentric) or occur systemically (multicentric). Lymph node abnormalities and organ dysfunction in Castleman disease are caused by hypersecretion of cytokines, commonly IL–6. The release of these cytokines is caused by HHV-8 (human herpesvirus; also known as Kaposi's sarcoma-associated herpesvirus (KSHV) that causes sarcoma Kaposi, commonly in AIDS patients, and primary effusion lymphoma) in HHV-8-associated MCD. Its MCD clinical features range from waxing and waning mild lymphadenopathy with B-symptoms to more severe cases involving intense inflammation, generalized lymphadenopathy, hepatosplenomegaly, vascular leak syndrome with anasarca, pleural effusions, ascites, organ failure, and even death. The most common ‘B Symptoms’ of MCD are high fevers, night sweats, weight loss, and loss of appetite. Laboratory findings commonly include low red cell count, low or high platelet counts, low albumin, high gamma globulin levels, elevated CRP levels, elevated ESR, IL-6, vascular endothelial growth factor (VEGF), and fibrinogen; positive ANA (anti-nuclear antibody), anti-erythrocyte autoantibodies, and anti-platelet antibodies; and proteinuria and polyclonal marrow plasmacytosis. Castleman disease is seen in POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes) and is implicated in 10% of cases of paraneoplastic pemphigus (a skin disease, usually blisters, related to cancer that triggers it as an autoimmune response).
· b) Some chemicals (e.g., polychlorinated biphenyls (PCBs), diphenylhydantoin, dioxin, and phenoxy herbicides).
· c) Medical treatment-related e.g., chemotherapy and radiotherapy.
· d) Genetic diseases (e.g., Klinefelter’s syndrome, Chediak – Higashi syndrome, ataxia-telangiectasia syndrome).
· e) Autoimmune diseases (Sjogren’s, celiac disease., rheumatoid arthritis, systemic lupus erythematosus (SLE)).
· Certain types of non-Hodgkin's lymphoma are defined as AIDS-defining cancers. Immune suppression rather than HIV itself is implicated in the pathogenesis of this malignancy.
· Treatment: combinations of chemotherapy, monoclonal antibodies (against CD20, e.g., rituximab)/ immunotherapy, radiotherapy, and hematopoietic stem cell transplantation.
· Prognosis: 5-year survival rates in the United States are 69%.
· Cutaneous T cell lymphoma (CTCL): a class of non–Hodgkin lymphoma. Unlike most non-Hodgkin's lymphomas, which are generally B cell-related, CTCL is caused by a mutation of T cells. The malignant T cells in the body initially migrate to the skin, causing various lesions that change shape as the disease progresses, typically beginning as an itchy rash and eventually forming plaques and tumors before metastasizing to other parts of the body.
· Of all cancers involving the same class of blood cells, 2% of cases are cutaneous T-cell lymphomas. There is some evidence of a relationship with HTLV (human T-lymphotropic virus).
· Classification: cutaneous T-cell lymphomas may be divided into the following types:
· Mycosis fungoides
· Pagetoid reticulosis
· Sezary syndrome
· Granulomatous slack skin
· Lymphomatoid papulosis
· Pituriasis lichenoides chronica
· Pityriasis lichenoides et varioliformis acuta
· CD30+ cutaneous CD30+ large lymphoma
· Non – mycosis fungoides CD30 – cutaneous large T – cell lymphoma
· Pleomorphic T–cell lymphoma
· Lannert Lymphoma
· Subcutaneous T–cell lymphoma
· Angiocentric lymphoma
· Blastic NK–cell lymphoma.
SYNDROMES RELATED TO HEMATOLOGICAL MALIGNANCIES
· Sweet's syndrome (SS; acute febrile neutrophilic dermatosis): a skin disease characterized by the sudden onset of fever, elevated WBCs, and tender red, well-demarcated papules and plaques that show dense infiltrate by neutrophils on histological examination.
· Classification: SS can be classified based on the clinical setting in which it occurs:
· a) Classical or idiopathic SS
· b) Malignancy – associated SS
· c) Drug-induced SS.
· Approximately 20% of cases are associated with malignancy, predominantly hematological, especially acute myelogenous leukemia (AML).
· An underlying condition (streptococcal infection, inflammatory bowel disease (IBD), non-lymphocytic leukemia and other hematological malignancies, solid tumors, pregnancy, collagen/rheumatological diseases (Behcet’s syndrome, rheumatoid arthritis (RA), etc.) is found in up to 50% of cases.
· Attacks of SS may precede the hematologic diagnosis by 3 months to 6 years so close observation is needed. However, it may occur in the absence of another known disease.
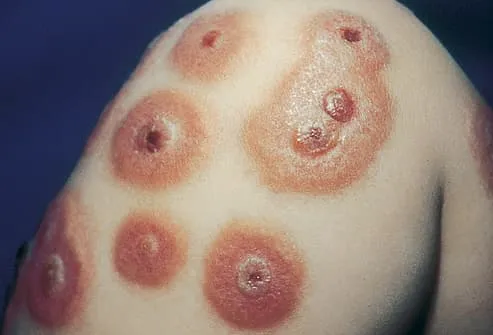{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· http://www.dermis.net/bilder/CD017/550px/img0083.jpg
· http://www.pcds.org.uk/ee/images/made/ee/images/uploads/clinical/Sweets_1__800_381_70_
{kind=link}
· http://www.pcds.org.uk/ee/images/made/ee/images/uploads/clinical/Sweets_1__800_381_70_
No comments:
Post a Comment