
Dr. James Manos (MD)
January 2, 2016
Tips in Hematology
Volume (2)
Volume (2)
2nd edition (revised)
CONTENTS
PERIPHERAL BLOOD SMEAR
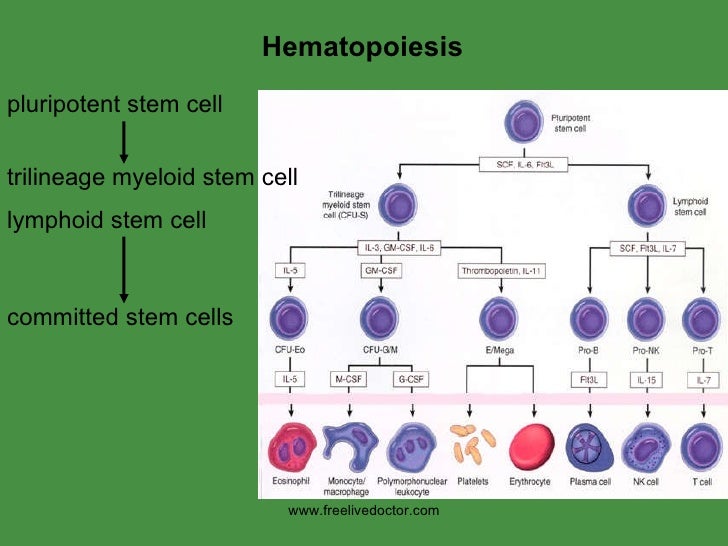
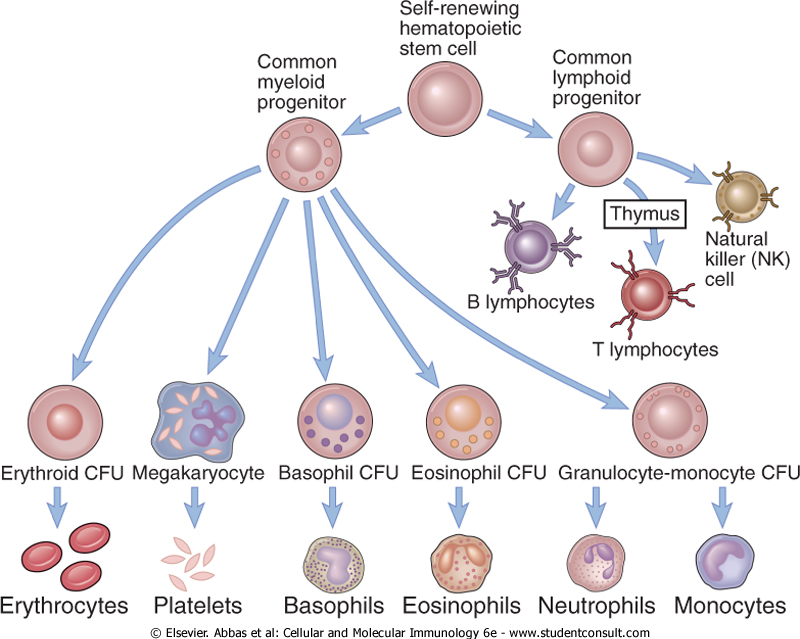
Hematopoiesis
When to check a peripheral blood smear (if not requested by the clinician) after having a CBC (FBC)
Red Blood cells (RBCs)
Normal red blood cells (discocytes)
Hypochromasia
Polychromasia – polychromatophilic erythrocyte
Reticulocytes
Microcytes
Macrocytosis
Round macrocytes
Oval macrocytes
Elliptocytes
Target cells (codocytes or leptocytes)
Stomatocytes
Spherocytes
Sickle cells (drepanocytes)
Teardrop-shaped red cells (dacryocytes)
Acanthocytes (spur cells or thorn cells or spiculated cells)
Bite cells (dangocytes or degmacytes), blister cells (pycnocytes) & ghost cells
Heinz bodies
Howell – Jolly bodies
Cabot rings
Schistocytes (fragment cells; sometimes referred to as ‘bite cells’)
Echinocytes (burr or crenated cells)
Dimorphic red cells
Basophilic stippling
Schuffner's granules
Nucleated red blood cells (NRBCs or normoblasts)
Polychromatophilic erythrocyte
Sideroblasts
Rouleaux formation
Pappenheimer bodies
Red blood cells (RBCs) & Parasites
Malaria
Babesia
White blood cells (WBCs)
Immature white blood cells (WBCs)
Granulocyte line immature cells (precursors)
Myeloblasts
Promyelocytes
Myelocytes
Metamyelocytes
Monocyte line immature cells (precursors)
Monoblasts
Lymphocyte line immature cells
Lymphoblasts
White blood cells in the peripheral blood
Atypical (reactive) lymphocytes
Atypical (reactive) lymphocytes in infectious mononucleosis
Large Granular Lymphocytes (LGL)
Pelger – Huet anomaly
Dohle bodies
Chediak–Higashi syndrome
Hypersegmented neutrophils
Toxic granulation of the neutrophils
Vacuolization of neutrophils
Bands/ immature neutrophils
SIRS (Systemic Inflammatory Response Syndrome)
Associated with hematological malignancies
Smudge cells (also called sometimes ‘basket cells’)
Auer rods
Hairy cells
Plasma cells
Leukemias
Αcute lymphoblastic leukemia (ALL; also known as acute lymphocytic leukemia and acute lymphoid leukemia)
Acute myeloid leukemia (AML, also known as acute myelogenous leukemia)
Chronic myelogenous leukemia (CML; also called chronic myeloid or myelocytic or myelogenic leukemia and chronic granulocytic leukemia (CGL))
Chronic lymphocytic leukemia (CLL, also known as chronic lymphoid leukemia)
Hairy cell leukemia (HCL, also called leukemic reticuloendotheliosis)
Platelets (Plts)
Clumping of platelets and falsely low platelet count (pseudo-thrombocytopenia)
Giant platelets
Platelet satellitosis
Parasites
Bacteria
Fungi/ Yeasts
INFECTIONS WITH HEMATOLOGICAL MANIFESTATIONS
Leishmaniasis
Typhoid fever (enteric fever)
Brucellosis
Pertussis (whooping cough)
GRANULOCYTE DISORDERS
Granulocytes
Azurophil granules
Agranulocytosis
Neutrophilia
Shift to the left
Neutropenia
SYNDROMES ASSOCIATED WITH NEUTROPENIA & RECURRENT INFECTIONS / IMMUNODEFICIENCY-RELATED SYNDROMES
Felty's syndrome
Chediak–Higashi syndrome
Wiskott–Aldrich syndrome (WAS)
HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS
GRANULOCYTE DISORDERS
HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS
GRANULOCYTE DISORDERS
PERIPHERAL BLOOD SMEAR
Hematopoiesis
· See:
{kind=link}
{kind=link}
{kind=link}
_diagram.png){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· When to ask for a peripheral blood smear (if not requested by the clinician) after having a CBC (FBC):
· In case a CBC (FBC; full blood count) is suggestive of an abnormal condition. Each hematologist has their own ‘guidelines.’ The author of this text personally (so the viewer of this text is not obligated to follow these empirical ‘recommendations’, but use published guidelines) will ask a blood film on a hospital patient in case a complete/full blood count (CBC; FBC) shows WBCs > 20 x 103/μL on a general medicine patient or > 25 x 103/μL on a surgical patient; if WBCs are low; if platelets are < 140,000 cells/microL or decrease substantially or increase (especially if > 600,000 cells/microL); and also if there is absolute lymphocytosis with absolute lymphocyte count (in adults) > 4,200 per microliter [other may ask a blood film if it is >5,000 per microliter; on the blood film the examiner will check especially if there are any reactive lymphocytes or lymphocytes and smudge cells indicative of CLL (chronic lymphocytic leukemia)] with or without inversion of the ratio of lymphocytes to neutrophils (some may not ask a blood film if the reversal of the ratio is not accompanied with absolute lymphocytosis, but the author of this text always asks a blood film in case of reversal, especially with absolute lymphocytosis more than 4-5) – in this case an old CBC (FBC) will help on checking if this inversion was long lasting. CLL and spleen lymphoma are in the differential diagnosis.
Red Blood cells (RBCs)
· Normal red blood cells (discocytes): normal red cells are biconcave discs. They have a mean diameter of 7µm, or approximately the size of a small lymphocyte nucleus. The red cell membrane is highly elastic and deformable, readily resuming its discoidal shape after the cessation of a deforming force.
· HypErchromasia: the RBCs are darker in color than usual. Causes: presence of spherocytes (hereditary spherocytosis), immune hemolysis, severe burns, microspherocytes in microangiopathic hemolytic anemia, macrocytosis (B12 or folate deficiency), viral infections (HIV, hepatitis).
· Polychromasia – polychromatophilic erythrocyte: Polychromatophilia may be defined as increased numbers of immature peripheral red blood cells that have a blue-gray tint on Wright-stained smears, indicating the presence of cytoplasmic RNA. These blue-staining RBCs are immature due to early release from bone marrow. They are usually larger than average. Many of these cells are proven to be reticulocytes when stained with supravital dyes such as brilliant cresyl blue. They appear under conditions of accelerated red cell production. Polychromasia (also known as polychromatophilia) is a disorder characterized by an abnormally high number of red blood cells in the bloodstream due to premature release from the bone marrow during blood formation. Polychromasia is usually a sign of bone marrow stress and immature red blood cells. 3 types are recognized, with types (1) and (2) being referred to as 'young red blood cells' and type (3) as 'old red blood cells.' Giemsa stain is used to distinguish all three types of blood smears. The young cells will stain gray or blue in the cytoplasm. These immature red blood cells are commonly called reticulocytes. All polychromatophilic cells are reticulocytes. However, not all polychromatophilic cells are reticulocytes.
· Causes of polychromasia:
· a) Associated with anemia: normocytic anemia is the most common type of anemia. This type of anemia is usually caused by underproduction of blood cells and hemolysis. Anemia can result from overproduction or underproduction of red blood cells, as well as from defective red blood cells. Because more red blood cells are needed in the body at that moment, they are released prematurely, leading to polychromasia.
· b) Assοciated with reticulocytosis: there is a slight correlation between polychromasia and reticulocytosis. Testing for polychromasia in blood cells is much easier than performing special staining for reticulocytosis. If polychromasia is found in blood cells, the reticulocyte count is measured to detect underlying disease or stress. If a small number of reticulocytes is found, it usually indicates bone marrow stress. If a high reticulocyte count is found, it typically indicates hemolysis, but a Coombs test may be performed to rule out immune-mediated hemolysis. Polychromasia can also be seen in blood smears when there is a normal reticulocyte count in case of infiltration of the bone marrow due to tumors, as well as fibrosis, or scarring, of the marrow.
· Blood film:
{kind=link}
{kind=link}
· Supravital stain:
{kind=link}
{kind=link}
· Reticulocytes: immature RBCs that are usually a blue–staining (polychromatic) color. A few are normal in circulation. Causes of elevated numbers: acute blood loss, hypoia, RBC destruction, sickle cell disease, G6PD deficiency, and autoimmune hemolytic anemias.
· Microcytes: are red cells with a diminished diameter, less than 6µm, and reduced thickness. The hemoglobin concentration of microcytes is reduced and often concentrated in a peripheral ring while the cell center is pale. This pale center may be so enlarged that the cell resembles a leptocyte or a thin, flattened red cell. Microcytes are a characteristic feature of iron deficiency anemia and thalassemia.
· Macrocytosis: large RBCs. Causes: vitamin B12 deficiency/ megaloblastic anemia (e.g. pernicious anemia), chronic alcoholism, MDS (myelodysplastic syndrome), hemolysis/ hemolytic anemia (mild MCV increase), antifolate drugs (such as phenytoin), hydroxyurea (hydroxycarbamide; a chemotherapy drug used in myeloproliferative disorders), liver disease, and marathon running/ ‘runner’s macrocytosis’ (the impact forces from running can lead to red blood cell hemolysis and accelerate red blood cell production; thus, there may be hemolysis and increased MCV on runners).
· Round macrocytes are red cells with an increased diameter of more than eight μm. They maintain a normal discoid shape and have a well-defined central depression. Round macrocytes are characteristic of accelerated erythrocytopoiesis, alcoholism, and liver disease.
{kind=link}
· http://www.andrews.edu/~albert/assets/CLSC260%20Web%20pages/Unit%2005_files/slide0053_image221.jpg
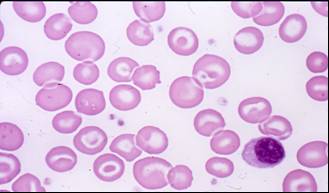{kind=link}
· Oval macrocytes are giant-sized red cells with a diameter of more than 9µm. Oval macrocytes are a characteristic feature of megaloblastic anemia.
{kind=link}
· http://www.andrews.edu/~albert/assets/CLSC260%20Web%20pages/Unit%2005_files/slide0054_image226.jpg
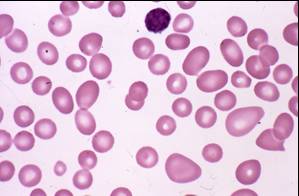{kind=link}
{kind=link}
· Elliptocytes are elliptical–shaped biconcave discs that vary in shape from slightly oval to cylindrical. They have both quantitative and qualitative abnormalities in two major proteins that comprise the membrane skeleton, namely spectrin and protein 4.1. While approximately 5% of elliptocytes are seen in normal blood films, between 30% and 100% are seen in hereditary elliptocytosis. They are also seen in anemias such as iron-deficiency anemia (but in this case, they don’t have spectrin or protein 4.1 abnormalities) and in myelofibrosis.
· Target cells (codocytes or leptocytes) are bell-shaped, thin-walled cells that have an increased surface area compared to volume. This results from red cell membrane expansion due to the accumulation of lecithin and cholesterol from free exchange with plasma lipids. The term "target" refers to the appearance of these cells when dried on a glass slide. They have a characteristic distribution of hemoglobin in the cell center and along the periphery, with the two regions separated by a pale ring. They resemble a bull’s eye. Target cells are associated with liver disease, hyposplenism, various anemias, and hemoglobinopathies such as sickle cell anemia, HbC disease, thalassemia, and some abnormal hemoglobins.
· Stomatocytes: are bowl-shaped red cells that exhibit a slit-like area of central pallor. Stomatocytes are associated with alcoholism, electrolyte imbalance, liver disease, and Rh null syndrome (a rare congenital hemolytic anemia in which there is no Rh in the blood). They are also a feature of two specific conditions: hereditary stomatocytosis and Southeast Asian ovalocytosis. In each of these conditions, the membrane defect is different. In hereditary stomatocytosis, there is a deficiency of protein 7.2b, or stomatin, in the red cell membrane. This leads to an influx of sodium ions into the red cell and an efflux of potassium ions from it. Hence, the red cells swell and are transformed from discocytes to bowl forms. They have a high mean cell volume (MCV), usually about 130 fl. In Southeast Asian ovalocytosis, increased ankyrin binding and decreased protein 3 mobility lead to the production of rigid red cells. These red cells are oval in shape, often with double transverse slits. They have a normal MCV. Stomatocytes may also occur as artifacts under abnormal physicochemical conditions in vitro.
· Spherocytes are red cells that are almost spherical in shape. They are no longer a biconcave disc. They appear as slightly smaller, hyperchromic red cells with little or no area of central pallor. The MCV of spherocytes is within the normal range. Spherocytes are a characteristic feature of hereditary spherocytosis and autoimmune hemolytic anemia.
· Large spherocytes (macro-spherocytes) are seen in hemolytic anemia. Small spherocytes (micro-spherocytes) are sometimes seen in severe burn cases.
· The spherocytes of hereditary spherocytosis are attributable to an intracorpuscular red cell membrane defect. Deficiency of spectrin, ankyrin, or band 3 protein leads to uncoupling of the skeletal lipid bilayer, resulting in membrane loss in microvesicles. The spherocytes of autoimmune hemolytic anemia are the end result of red cell coating with immunoglobulin (IgG) and/or complement proteins.
· Sickle cells (drepanocytes): biconcave discs that, upon deoxygenation, change shape to become crescent or sickle-shaped. They have abnormal hemoglobin, HbS, due to the replacement of glutamic acid with valine at the sixth position on the β chain. Sickle cells are deformed by the precipitation of polymerized HbS. They appear as sickles or crescent-shaped red cells on the blood film. They are a characteristic feature of sickle cell disease. Sickle cells are also seen in double-heterozygote disorders that include HbS.
· Teardrop-shaped red cells (dacryocytes) are characterized by a single elongated or pointed extremity. They are a characteristic feature of leucoerythroblastic disorders such as myelofibrosis, other myeloproliferative disorders, and metastatic carcinoma. Teardrops are also present in thalassemia, pernicious anemia/ megaloblastic anemia, MDS (myelodysplastic syndrome), myeloid metaplasia, and some hemolytic anemias.
· Acanthocytes (spur cells, thorn cells, or spiculated cells) are irregular, round, hyperchromic red cells with several fine, spine-like projections (5 – 10 spicules) spaced unevenly over their entire surface. Acanthocytes are a characteristic feature of abetalipoproteinemia, chronic alcoholism (with cirrhosis), liver disease, splenectomy, hemolytic anemia, and thalassemia.
· Other causes include the McLeod syndrome, chorea-acanthocytosis (a rare hereditary disease characterized by acanthocytes and neurodegeneration with neurological problems such as epilepsy, choreiform movement disorders, etc.), neuroacanthocytosis (acanthocytosis combined with neurological issues by a variety of syndromes that affect the brain & basal ganglia), anorexia nervosa, infantile pyknocytosis, hypothyroidism, alcoholism, idiopathic neonatal hepatitis, congestive splenomegaly, Zieve syndrome (an acute metabolic condition that can occur during withdrawal from prolonged alcohol abuse; defined by hemolytic anemia with acanthocytes, hyperlipoproteinemia, jaundice & abdominal pain) and chronic granulomatous disease.
· Note: McLeod syndrome is an X-linked recessive genetic disorder that may affect the blood, brain, peripheral nerves, muscle & heart. Patients usually notice symptoms in their 50s, and the course is generally slowly progressive. Typical features include peripheral neuropathy, cardiomyopathy, hemolytic anemia, acanthocytosis (on the peripheral blood film), chorea, facial tics, lip and tongue biting, seizures, behavioral changes, and late-onset dementia.
· Pyknocytes are RBCs that appear small, dark, distorted, irregular, without central pallor, with abnormal projections and would typically be identified on a peripheral blood smear. Infantile pyknocytosis is the most associated condition and is a rare pediatric hematological condition, and a potential cause of neonatal hemolytic anemia. Also causes are G6PD deficiency and pyruvate kinase deficiency.
· Bite cells (dangocytes or degmacytes), blister cells (hemighost cells) & ghost cells: Bite cells are formed when Heinz bodies (the product of oxidant stress on hemoglobin), together with some red cell content, are removed (by splenic macrophages) from red cells as they pass through the spleen. When the red cell membrane around the bite repairs, a blister-like structure forms, hence the term blister cell. Blister cells appear as RBCs containing peripherally located vacuole.
Bite & blister cells are characteristic features of oxidant hemolysis seen in subjects with normal or reduced G6PD levels after, e.g., administration of dapsone, salazopyrine, and some antimalarial medications. Bite cells appear in thalassemia, NADPH and G6PD deficiency or other RBCs enzymopathies.
· Blister cells:
{kind=link}
{kind=link}
· Bite cells:
· https://classconnection.s3.amazonaws.com/424/flashcards/1123424/jpeg/graphic080311333634735151.jpeg
{kind=link}
{kind=link}
· Ghost cells:
{kind=link}
{kind=link}
· Heinz bodies: inclusions within RBCs composed of denatured hemoglobin. They appear more clearly when supravirtually stained (such as with new methylene blue, bromocresol green, or crystal violet). Causes: NADPH deficiency (dysfunction in glutathione peroxidase), G6PD (glucose – 6 – phosphate dehydrogenase) deficiency (exacerbated by administration of oxidant drugs such as primaquine, dapsone, and quinidine), chronic liver disease, thalassemia, hyposplenism or asplenia, unstable hemoglobin variant, and autoimmune hemolytic anemia.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Howell-Jolly bodies: are spherical, blue-black inclusions of red blood cells seen on Wright-stained smears. They are nuclear fragments of condensed DNA, 1 to 2m in diameter, normally removed by the spleen. They are seen in severe hemolytic anemias, in patients with dysfunctional spleens/ hyposplenism or after splenectomy, and also in sickle cell anemia and megaloblastic anemia.
· Cabot rings are threadlike inclusions that form a ring within the RBC. The may form a single ring (loop) or a figure 8 (8-shaped loop). It may be seen in a variety of anemias, including megaloblastic anemia and myelodysplastic syndromes (MDS). Cabot rings should not be confused with malaria rings.
{kind=link}
{kind=link}
· Schistocytes (fragment cells; sometimes referred to as ‘bite cells’) are sharp, pointed, fragmented red cells produced by a microangiopathic process. Red cells are fractured or ripped as they pass across strands of fibrin in damaged vessels, for example, in microangiopathic hemolytic anemia, such as in hemolytic-uraemic syndrome (HUS), in thrombotic thrombocytopenic purpura (TTP; Moschcowitz syndrome), and DIC (disseminated intravascular coagulation), pro/eclampsia, malignant hypertension, or as they pass across a damaged or prosthetic heart valve. Other causes include severe burns, uremia, toxins, MDS (myelodysplastic syndrome), march hemoglobinuria, and hemolytic anemias caused by physical agents.
· Echinocytes (burr or crenated cells): are red cells with varying numbers of spines (10 – 30 blunt projections) occurring at irregular intervals on the cell membrane. Burr cells are produced due to damaged glomeruli in severe glomerulonephritis and renal failure/ uremia.
· Echinocytes, as an artifact, may result from faulty blood smear drying or exposure to hyperosmotic solutions. They are invariably present as artifacts that develop when blood is allowed to stand for a prolonged period at 20°C before the blood film is made.
· They are also present in dehydration resulting from severe gastroenteritis.
· Other causes include liver problems and electrolyte disorders.
· Dimorphic red cells: the presence of 2 different populations of RBCs (red blood cells), e.g., normocytic & macrocytic. It describes heterogeneity in red blood cell size, typically with two distinct populations. Causes: partially treated iron deficiency, mixed deficiency anemias (e.g., folate/B12 and iron together), red cell transfusion, sideroblastic anemia, post-gastrectomy, liver disease, iron deficiency, vitamin B12 and folate deficiency, initiation of iron treatment on sideropenic (iron deficiency) anemia, MDS (myelodysplastic syndrome), RARS (refractory anemia with ringed sideroblasts).
· Basophilic stippling: on the blood smear, the RBCs display small dots at the periphery that are ribosomes and can often be found in the peripheral blood smear, even in some healthy individuals. It is associated with several conditions, including myelodysplastic syndrome (MDS), lead poisoning (normocytic anemia), myelofibrosis, nutritional deficiencies, megaloblastic anemia, sideroblastic anemia, arsenic poisoning, thalassemia, HbH disease (a form of alpha thalassemia), hereditary pyrimidine 5’–nucleotidase deficiency, severe thalassemia, and thrombotic thrombocytopenic purpura (TTP).
· Schuffner's granules: may be found in cases of Plasmodium vivax. These granules appear as orange-to-pink-colored stippling throughout the red blood cell. They may not be visible when normal staining times are used. The smears should be allowed to stain for three hours to detect these granules.
· Nucleated red blood cells (NRBCs or normoblasts): represent the stages of a red blood cell before it matures. Cells of this stage are usually seen in newborn infants and in patients with hemolytic crises. The average size of the normoblast is 7 – 12 µm in diameter. The cytoplasm is pink. The nucleus is pyknotic (a homogeneous blue-black mass with no structure). Normally, NRBCs are only found in the circulation of fetuses and newborn infants. Nucleated RBCs can be normal in infants for a brief time after birth. After infancy, RBCs normally only contain a nucleus during the very early stages of the cell's life, and the nucleus is ejected before the cell is released into the bloodstream. Thus, if NRBCs are seen on an adult's peripheral blood smear, it suggests a very high demand for RBC production by the bone marrow and that immature RBCs are being released into the circulation. Possible pathologic causes include severe anemia, myelofibrosis, thalassemia, miliary tuberculosis, cancers involving bone marrow, and chronic hypoxemia. In chronic myelogenous leukemia (CML), occasional nucleated red blood cells are seen on the peripheral blood smear.
· See:
{kind=link}
· Sideroblasts: Iron granules may be seen when RBCs are stained with Prussian blue dye. Sideroblasts are immature siderocytes and may form a ring pattern (ring sideroblasts) indicative of sideroblastic anemia.
· Rouleaux formation occurs when red blood cells form stacks or rolls. They occur when plasma protein concentration is high, and as a result, the ESR is also increased. Acute–phase proteins, especially fibrinogen, interact with sialic acid on the surface of RBCs to facilitate the formation of rouleaux. An increase in the ratio of RBCs to plasma volume, as seen in anemia and hypovolemia, increases rouleaux formation and accelerates sedimentation. Rouleaux formation is retarded by albumin proteins. Rouleaux formation is due either to an artifact (such as not preparing the blood smear soon enough after placing the blood on the slide) or to high concentrations of abnormal globulins or fibrinogen. It is found in multiple myeloma and macroglobulinemia, and also in infections, inflammatory & connective tissue disorders, cancer, and diabetes mellitus.
{kind=link}
· Pappenheimer bodies are iron-containing granules in red blood cells that are seen because the iron is aggregated with mitochondria and ribosomes. They appear as faint violet or magenta specks, often in small clusters, due to the staining of the associated protein. They are associated with severe anemias and thalassemias.
Red blood cells (RBCs) & Parasites
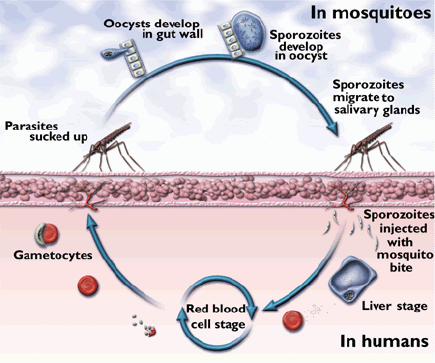
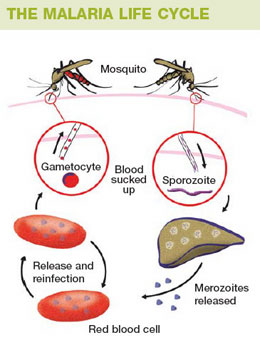
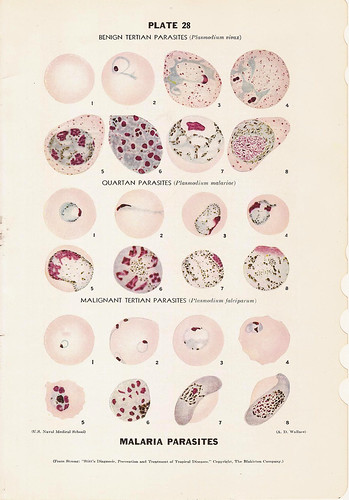
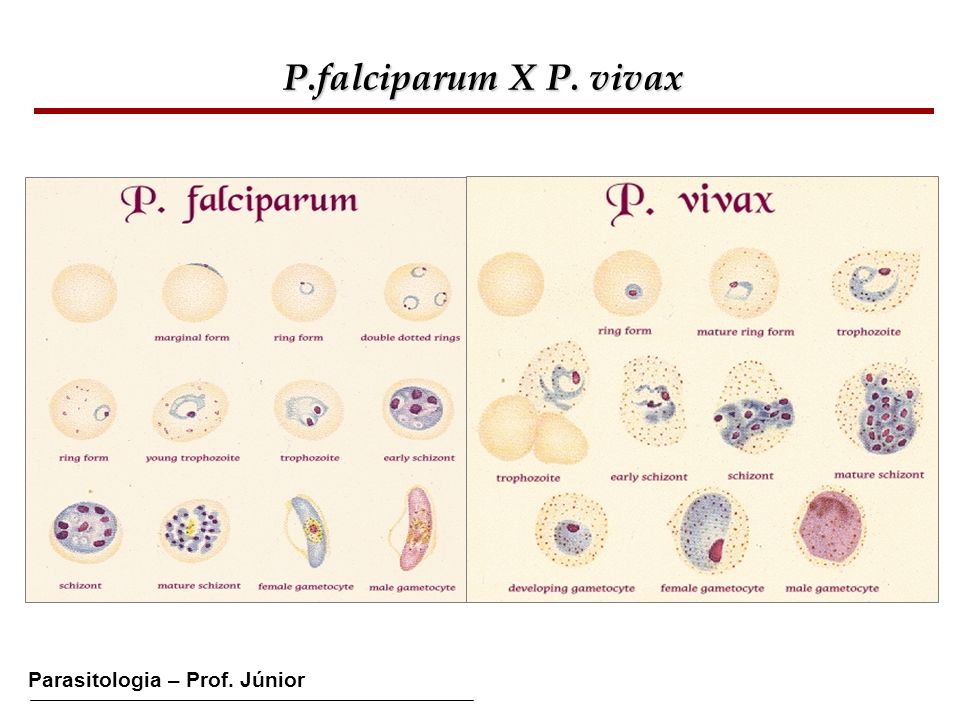
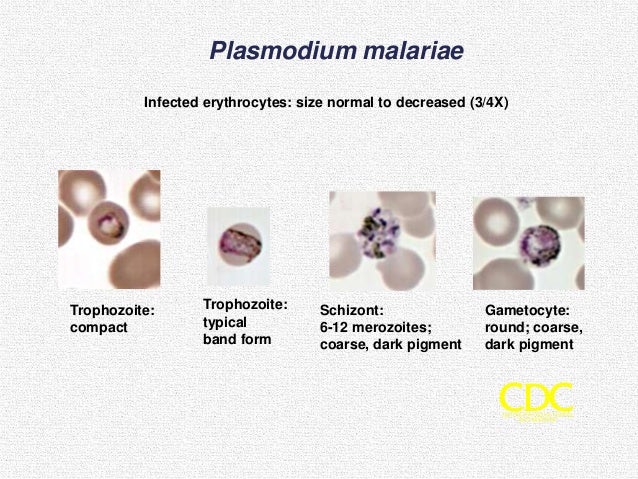
· Malaria is a disease caused by the parasite Plasmodium. The four species of Plasmodium most commonly found in man are vivax, malaria, falciparum, and ovale. Malaria is mainly transmitted from person to person through the bite of the female Anopheles mosquito. Other means of transmission are through the use of contaminated needles, congenital means, and blood transfusions. When the infected Anopheles mosquito bites a human, sporozoites are injected into the individual's peripheral blood. The sporozoites then invade the liver. When the merozoite penetrates the red blood cell, the parasite develops into the ring form of the trophozoite and then into a mature schizont. The merozoites rupture from the mature schizonts and penetrate other red blood cells. Fever and chills are associated with red blood cell rupture. Malarial parasites should be sought in all patients with suspected intravascular hemolysis.
· A negative for malaria blood film should be repeated 3 times to exclude malaria (other tests, such as a malaria antigen test, may be used). A case study concluded that it is crucial to consider P. falciparum malaria in the differential diagnosis for those with malaria exposure in the last 12 months, and to always take three blood films if there is any clinical suspicion to make a diagnosis of malaria. Lymphopenia and thrombocytopenia may indicate malaria (the sensitivity of thrombocytopenia in malaria is 60%, and the specificity is 95%). A study in Nigeria found that headache, fever, chills, and rigors were the most common malaria symptoms in adults.
· Reference: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1420790/
· Thick & thin smears: a) Thick smears. Thick smears consist of a thick layer of de-hemoglobinized (lysed) red blood cells. The blood elements (including parasites, if any) are more concentrated (app. 30×) than in an equal area of a thin smear. Thus, thick smears allow more efficient detection of parasites (increased sensitivity). However, they do not permit an optimal review of parasite morphology. For example, they are often not adequate for species identification of malaria parasites: if the thick smear is positive for malaria parasites, the thin smear should be used for species identification. b) Thin smears consist of blood spread in a layer such that the thickness decreases progressively toward the feathered edge. In the feathered edge, the cells should be in a monolayer, not touching one another.
· See:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
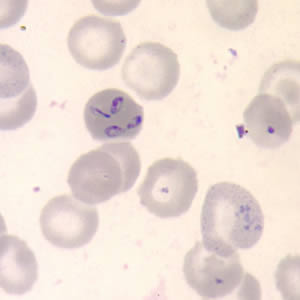
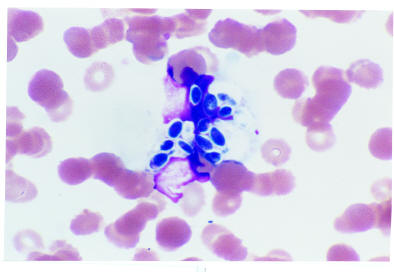
· Babesia are small ringlike protozoa within erythrocytes that resemble the ring stages of falciparum malaria. But it is far more rare than malaria. Most infections result from Babesia microti, transmitted from wild feral deer mice to humans by the tick Ixodes dammini. It occurs in Nantucket Island, in coastal regions of the northeastern US, and in California. It has been found in France, Ireland, Scotland, and other European countries. Babesia may be rarely transmitted from blood transfusion. The diagnosis of babesiosis can be made by demonstrating ring-shaped parasites on Wright-Giemsa-stained smears. Babesia appears as tiny rings with a minute chromatin dot and minimal cytoplasm. They may be round, oval, elongated, or ameboid. One or two chromatin dots, which stain dark purple, may be observed. More than one ring can be seen in an RBC. Tetrad forms may be observed and aid in the positive identification of Babesia.
· Peripheral blood smear: Babesia inclusions: both thick and thin blood smears should be prepared. The parasites appear as pleomorphic, ringlike structures (ring forms). They can resemble the early forms of malarial parasites, particularly Plasmodium falciparum. However, falciparum rings are rarely extracellular; Babesia parasites tend to be much more pleomorphic and may have 5–6 rings per cell, vs. the common 2 in falciparum cases. The classic arrangement of the 4 rings, called the Maltese cross, is not always seen. One negative blood smear does not rule out a Babesia infection.
· See:
{kind=link}
{kind=link}
White blood cells (WBCs)
· Immature white blood cells (WBCs)
· See also:
· Granulocyte line immature cells (precursors)
· Myeloblasts are the first stage of the granulocytic series that is identifiable by light microscopy. It may be difficult to distinguish myeloblasts from other blasts in the peripheral blood unless one uses special stains or infers their identity from the presence of other immature cells of the same line. A myeloblast can be distinguished from a promyelocyte by its lack of cytoplasmic granulation. The nucleus occupies most of the cell with a nucleus-cytoplasm ratio of 6:1. The nucleus is composed of very fine, non-aggregated chromatin that stains light blue to reddish-purple with Wright's stain. Two to five distinct nucleoli are usually present. The nucleus is often bordered on one side by a distinct perinuclear zone.
· See:
{kind=link}
{kind=link}
{kind=link}
· Promyelocytes are the second largest stage in the granulocytic series. The large, reddish-purple granules that characterize promyelocytes are nonspecific in that they are shared by the other granulocytes (eosinophils and basophils). These granules are peroxidase-positive, and a lipid component reacts with the Sudan black stain, providing a second cytochemical reaction for identifying large mononuclear cells in blood and bone marrow smears. The cell size, 12 to 20 µm, is the same as that of a myeloblast, but the nucleus-to-cytoplasm ratio is less, usually from 3:1 to 2:1. The chromatin is still fine, but some aggregation is evident. One to three nucleoli are also visible, although these can be obscured by heavy granulation.
· See:
{kind=link}
{kind=link}
{kind=link}
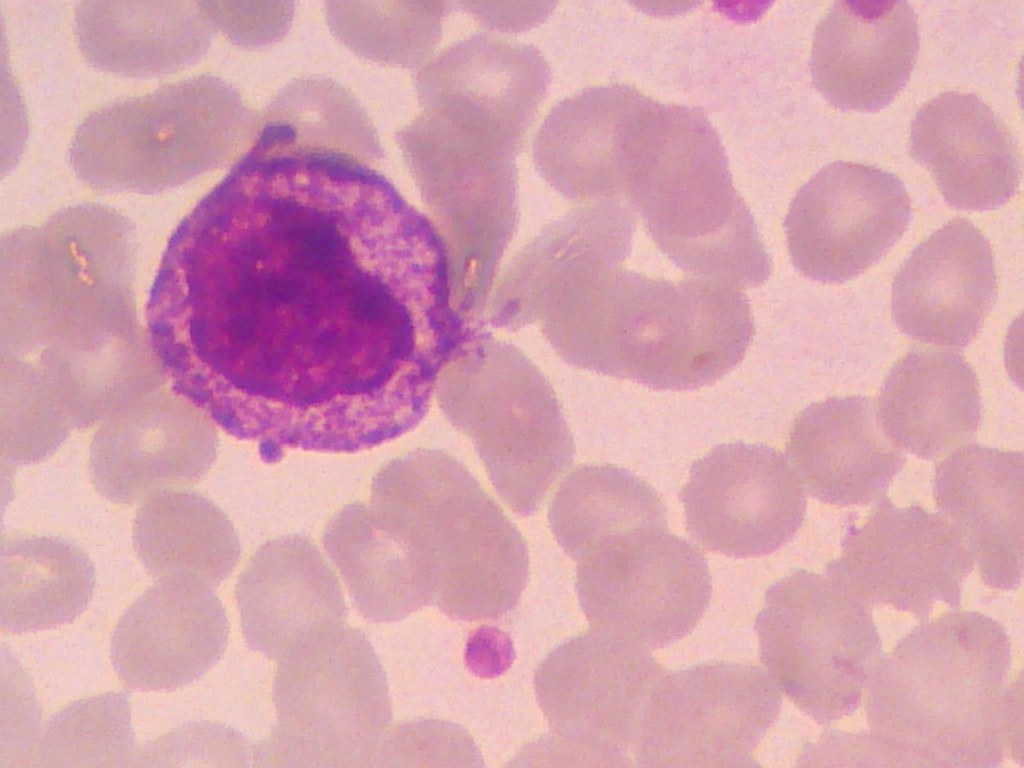
· Myelocytes: have a cell diameter from 10 to 18 µm. The nucleus-to-cytoplasm ratio is 1:1, and the oval or round nucleus is often eccentrically located. Chromatin is finely granulated in early myelocytes and more aggregated in later cells.
· See:
{kind=link}
{kind=link}
{kind=link}
· Metamyelocytes: have a cell diameter from 10 to 18 µm, and the nucleus-to-cytoplasm ratio is 1:1. Indentation of the nucleus begins at this stage, forming an outline that varies from slightly kidney-shaped to that of a broad V shape.
· See:
· https://classconnection.s3.amazonaws.com/109/flashcards/1951109/png/metamyelocyte1348965097254.png
{kind=link}
{kind=link}
· Monocyte line immature cells (precursors)
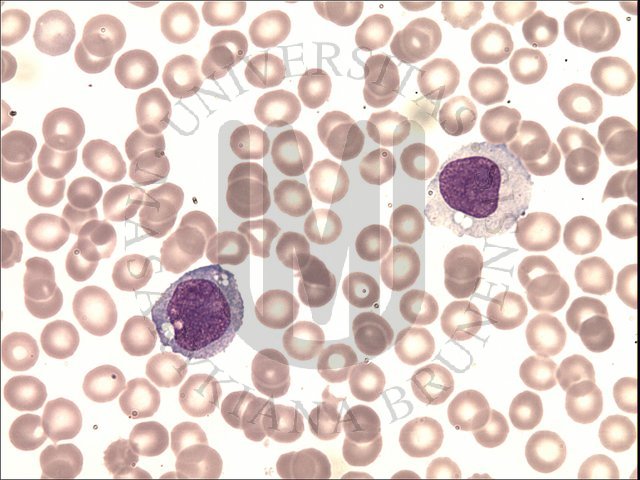
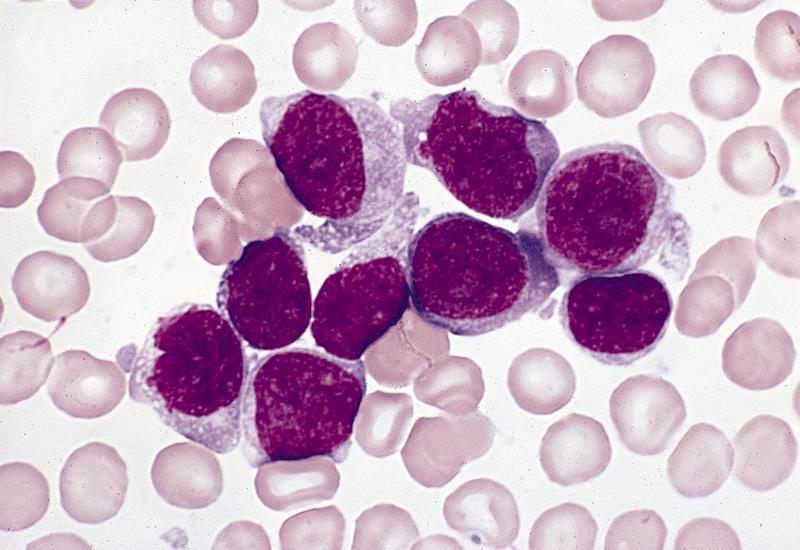
· Monoblasts are the first stage of monocyte-macrophage maturation. A monoblast is about 12 to 20 µm in diameter, has a nuclear to cytoplasm ratio of 4:1 to 3:1, and, like most myeloid blasts, has a round to oval nucleus with fine, lightly dispersed chromatin. From one to four nucleoli may be visible. The nucleus may be either central or eccentric, and it may show evidence of indentation or folding. The cytoplasm is agranular, stains moderately to lightly basophilic, and often has an intensely stained periphery and a prominent perinuclear zone. Monoblasts never appear in the normal peripheral blood.
· See:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Lymphocyte line immature cells
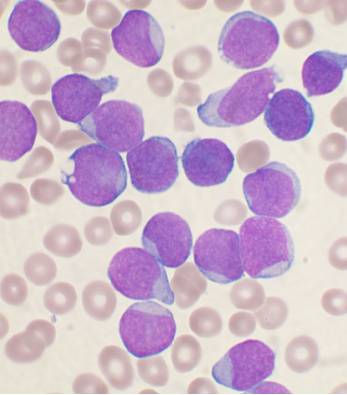
· Lymphoblasts: are 12 – 20 µm in diameter with a round to oval nucleus, sometimes eccentric in location. The nucleus to cytoplasm ratio is about 4:1, and the periphery of both the nucleus and the cell may be irregular in outline. The fine, highly dispersed nuclear chromatin stains a light reddish purple, and one or two pale blue or colorless large nucleoli are visible. The cytoplasm is usually agranular and deeply to moderately basophilic, with marginal (peripheral) intensity a common characteristic.
· See:
{kind=link}
{kind=link}
{kind=link}
Flower-like lymphocytes. Peripheral blood smear may show flower-like lymphocytes. This may be from Adult T-Cell Leukemia/Lymphoma (ATLL; it is caused by a retrovirus called the human T-cell leukemia virus (HTLV1). However, the evaluation of flower-like lymphocytes must be performed carefully because these cells are not only found in ATLL patients but also in patients with various other diseases involving reactive or neoplastic lesions, or even in healthy individuals. They may be found in the peripheral blood during routine daily blood examinations, such as those of patients with hepatitis B or anemia. Flower-like lymphocytes should also be considered for the differential diagnosis of flower-like plasma cells. In Sezary syndrome, lymphocytes may have a flower-like or cerebriform nuclear shape in the peripheral blood. For flower-like lymphocytes, see https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9160704/ and https://imagebank.hematology.org/image/12563/flower-cells-of-leukemia
· White blood cells in the peripheral blood
· Atypical (reactive) lymphocytes: lymphocytes that, as a result of antigen stimulation, have become quite large, sometimes more than 30 µm in diameter. The cells vary greatly in size and shape. The nucleus is less clumped than that of the normal lymphocyte. The shape of the nucleus ranges from elliptic to cleft to folded. The chromatin patterns appear similar to those of a blast, and faintly stained multiple nucleoli are visible. The cytoplasm may range from large, deeply basophilic, and abundant to unevenly stained and granular.
· Causes of reactive lymphocytosis include infections from Epstein-Barr virus (EBV), cytomegalovirus (CMV) that cause infectious mononucleosis (CMV causes the minority of the cases); infections from rubella, roseola, syphilis, toxoplasmosis, and viral hepatitis (e.g., hepatitis C), group B streptococci (GBS, Streptococcus agalactiae), and hantavirus. Other causes include drugs (such as phenytoin), radiation, hormonal causes (e.g., Addison’s disease), autoimmune diseases (e.g., rheumatoid arthritis (RA)), vaccination, and lymphoproliferative disorders (e.g., common variable immune deficiency, Chediak-Higashi syndrome, Wiskott-Aldrich syndrome, X-linked lymphoproliferative disorders).
{kind=link}
![http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/reactive_lymphocyte/reactive-atypical-lymphocytes-[2-bl034-2].jpeg?Width=600&Height=450&Format=4](http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/reactive_lymphocyte/reactive-atypical-lymphocytes-%5b2-bl034-2%5d.jpeg?Width=600&Height=450&Format=4){kind=link}
{kind=link}
{kind=link}
· Atypical (reactive) lymphocytes in infectious mononucleosis may be because of the Epstein-Barr virus (EBV), a DNA herpes-type virus that infects B lymphocytes. Also, Cytomegalovirus (CMV) causes a minority of the cases of infectious mononucleosis. Patients present with mild to severe adenopathy, hepatosplenomegaly, fever, malaise, pharyngitis, and a characteristic peripheral blood smear demonstrating reactive lymphocytes.
· Atypical (reactive) lymphocytes in infectious mononucleosis: reactive lymphocytes may resemble monocytes or immature cells and are highly pleomorphic. Several classification schemes are used to categorize reactive lymphocytic forms (Downey, Wood, and Frenkel, etc.). One type of cell (Downey type I) is small and has indented or lobulated nuclei. The chromatin is clumped. Nucleoli, if visible, are small, and the cytoplasm is scant and basophilic with numerous small vacuoles present. Azurophilic granules can also be seen on occasion. Another type of reactive lymphocyte resembles a large lymphocyte and corresponds to a Downey type II cell. These cells have a round-to-oval nucleus, moderately clumped chromatin, and absent or indistinct nucleoli. They contain abundant grey-blue cytoplasm that frequently partially surrounds adjacent red cells and has a darker staining furled margin (“pencil lining effect”). Occasionally, basophilia radiating out from the nucleus is also present, known as “radial bluing.” Immunoblasts and immunoblastic-like reactive lymphocytes (Downey type III) are larger cells with round-to-oval nuclei, moderately dispersed chromatin, and one or more prominent nucleoli. These transformed cells may resemble lymphoma cells or blasts. Their cytoplasm is moderately abundant and stains deeply basophilic. The nucleus-to-cytoplasm ratio is high (3:1 or 2:1). Plasmacytoid reactive lymphocytes resemble plasma cells and are intermediate in size, with a round-to-oblong shape. They have round nuclei that may be centrally placed or highly eccentric. Nucleoli are generally not visible, and the cytoplasm is moderately abundant, homogeneous, light blue to deep slate blue, and shows a clear paranuclear zone. While it may often be difficult to distinguish reactive lymphocytes from lymphoma cells, lymphoblasts, and prolymphocytes, the key distinguishing feature of reactive lymphocytes is their wide range of morphologic appearance within the same blood film. In contrast, lymphoma cases tend to show a monotonous population of abnormal cells.
· Diagnosis of infectious mononucleosis involves identifying atypical lymphocytes in peripheral blood smears. During acute EBV disease, the number of lymphocytes increases to 50 – 60% of the total leukocytes in the peripheral blood (a count of 20 000–50 000/ml), of which 10% are atypical lymphocytes (95% are T lymphocytes, 5% are B lymphocytes), or Downey cells.
· Hoagland’s criteria for the diagnosis of infectious mononucleosis are the most widely cited: at least 50% lymphocytes and at least 10% atypical lymphocytes in the presence of fever, pharyngitis, and adenopathy, and confirmed by a positive serologic test. While quite specific, these criteria are not highly sensitive and are most useful for research purposes. Only about one-half of patients with symptoms suggestive of infectious mononucleosis and a positive heterophile antibody test meet all of Hoagland’s criteria.
· The presence of atypical lymphocytes is probably the earliest indication of EBV infection but is not specific to EBV infection (see above). Modest leukocytosis is observed, and an elevated erythrocyte sedimentation rate (ESR) is also frequently reported.
· See:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Large Granular Lymphocytes (LGL): The cytotoxic response to a specific antigen is mediated by two highly professional, although extremely different players, cytotoxic T lymphocytes and natural killer (NK) cells, with the common feature of a morphological appearance of large granular lymphocytes (LGL): both are lymphocytes with coarse azurophilic granules within their cytoplasm, containing the cytotoxic weapons they are equipped with. The most striking difference is the way in which they recognize targets: T-cell or NK-cell receptors. Large granular lymphocytes (LGL) are defined as nonadherent mononuclear cells with cytoplasmic azurophilic granules, avid receptors for the Fc portion of IgG, and cytotoxic functions [NK or ADCC (antibody-dependent cell-mediated cytotoxicity) activities]. Proliferations of LGL comprise a spectrum of conditions, ranging from polyclonal, usually self-limited expansions to asymptomatic clonal LGL expansions to clearly symptomatic leukemic disease. Polyclonal expansions of LGL are usually transient due to a viral infection, such as Epstein-Barr virus (EBV) or cytomegalovirus (CMV), neoplasm, or autoimmune diseases; these disorders sometimes develop after splenectomy. In contrast, clonal LGL proliferations are stable for a while, whether or not the patients are symptomatic. In normal individuals, only a minority of circulating blood lymphocytes display the morphological characteristics of LGL. Most of these lymphocytes have a cytotoxic T-cell phenotype (CD3+ CD8+ CD4−), while only a few are NK cells (CD3− CD16+). Clonal expansions of LGL may comprise both T- and NK-cell-derived diseases, but the WHO considers NK-cell-derived diseases as a separate group referred to as chronic lymphoid disorders of NK cells. T-cell LGL (T-LGL) leukemia represents the clonal expansion of a terminally differentiated cytotoxic T-cell bearing a fully functional α/β+ T-cell receptor (TCR) with the corresponding CD8+ CD4− or, rarely, CD4+ CD8−/+dim phenotypic pattern; expansions of γ/δ+ TCR cells account for only rare cases. Less information is available for NK cells in chronic lymphoproliferative disorders (CLPD), although it seems that these cells are activated in vivo by persistent/active infection/stimulation.
{kind=link}
{kind=link}
{kind=link}
![http://www.pathpedia.com/education/eatlas/histology/blood_cells/large-granular-lymphocyte-(lgl)-[ce05h-1].jpeg?Width=600&Height=450&Format=4](http://www.pathpedia.com/education/eatlas/histology/blood_cells/large-granular-lymphocyte-(lgl)-%5bce05h-1%5d.jpeg?Width=600&Height=450&Format=4){kind=link}
{kind=link}
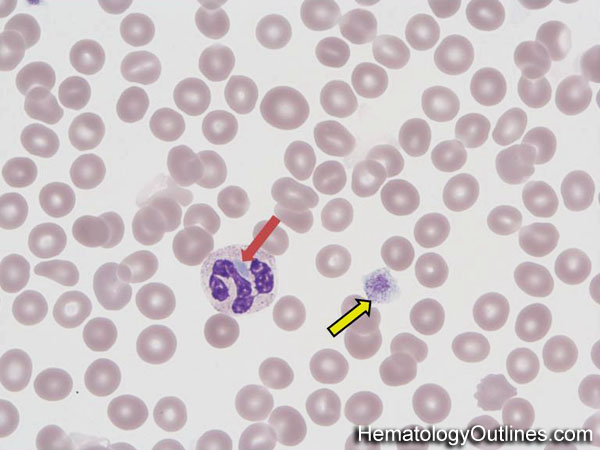
· Pelger – Huet anomaly is a benign hereditary condition characterized by decreased segmentation in the neutrophils. These neutrophils usually contain two lobes but appear to function normally. It is a blood laminopathy (associated with the lamin B receptor) characterized by neutrophils with a hypopigmented nucleus. It is a genetic disorder with an autosomal dominant inheritance pattern.
· Anomalies resembling Pelger–Huet anomalies, but acquired, have been described as pseudo-Pelger–-Huet anomalies. These can develop during AML (acute myelogenous leukemia), CML (chronic myelogenous leukemia), and MDS (myelodysplastic syndrome). In patients with these conditions, pseudo–Pelger–Huet cells tend to appear late in the disease, often after considerable chemotherapy has been administered. This morphology may also occur in multiple myeloma and in leukemoid reactions secondary to bone marrow metastases. The morphologic changes have also been described in myxedema associated with panhypopituitarism; vitamin B12 and folate deficiency; enteroviral infection; malaria; muscular dystrophy; drug sensitivity; sulfa–drugs and valproate (an antiepileptic) toxicities, etc.
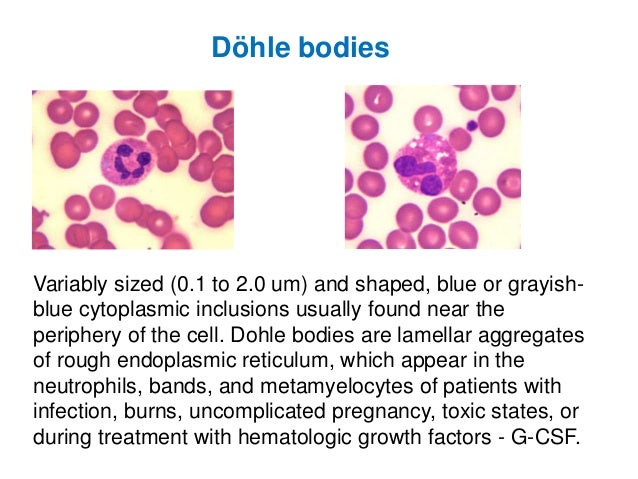
· Dohle bodies: appear as a small, light blue-gray staining area in the cytoplasm of the neutrophil. They are found in poisoning, burns, infections, and following chemotherapy.
{kind=link}
· Note: Dohle–like bodies may appear in blood film on patients with May–Hegglin anomaly, a rare genetic disorder characterized by thrombocytopenia, pale-blue leukocyte inclusions (Dohle–like bodies), and giant platelets.
{kind=link}
{kind=link}
{kind=link}
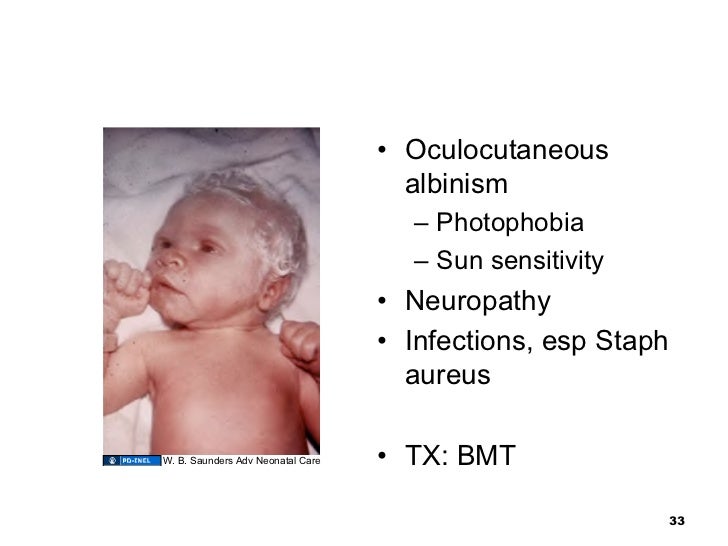
· Chediak–Higashi syndrome: a rare autosomal recessive disorder that arises from a mutation of a lysosomal trafficking regulator protein, which leads to a decrease in phagocytosis that results in recurrent pyogenic infections, partial ocular-cutaneous albinism, and peripheral neuropathy.
· The diagnosis is confirmed by bone marrow smears that show "giant inclusion bodies" in the cells that develop into white blood cells (leukocyte precursor cells).
![http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/chediak-higashi_syndrome/chediak-higashi-syndrome-[4-bl050-4].jpeg?Width=600&Height=450&Format=4](http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/chediak-higashi_syndrome/chediak-higashi-syndrome-%5b4-bl050-4%5d.jpeg?Width=600&Height=450&Format=4){kind=link}
· http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/chediak-higashi_syndrome/chediak-higashi-syndrome-[3-bl050-3].jpeg?Width=600&Height=450&Format=4http://image.slidesharecdn.com/011209-jlevine-myeloidcelldisorders-110727140520-phpapp01/95/011209-myeloid-cell-disorders-33-728.jpg?cb=1311775763
![http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/chediak-higashi_syndrome/chediak-higashi-syndrome-[3-bl050-3].jpeg?Width=600&Height=450&Format=4](http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/chediak-higashi_syndrome/chediak-higashi-syndrome-%5b3-bl050-3%5d.jpeg?Width=600&Height=450&Format=4){kind=link}
{kind=link}
· Hypersegmented neutrophils are neutrophils with five or more nuclear lobes. They are seen in disorders of nuclear maturation, such as megaloblastic anemias.
· There are some conditions in which hypersegmented neutrophils may be seen, such as megaloblastic anemias (including folic acid deficiency and pernicious anemia). Individuals receiving chemotherapy or having long-term chronic infections may also have hypersegmented neutrophils. However, in seeming contradiction to this, some studies have strongly associated neutrophil hypersegmentation with iron deficiency anemia.
· Toxic granulation of the neutrophils appears as dark blue-black granules in the cytoplasm of neutrophils. These granules are thought to be primary granules. Artifactual heavy granulation caused by poor staining is seen evenly spread throughout each cell and in all granulocytes, whereas toxic granulation is unevenly spread throughout the cytoplasm of specific cells. Large amounts of toxic granulation can give the neutrophil a bluish appearance. Conditions associated with toxic granulation include severe infectious processes or sepsis, severe inflammatory states, trauma, burns, certain cytokine medications, pregnancy, bone marrow response to myelosuppressive therapy, drug poisoning, and uremia.
· See:
· Vacuolization of neutrophils: vacuoles are clear, unstained areas in the cytoplasm of neutrophils. On vacuolization, vacuoles appear as holes in the cytoplasm and are frequently associated with toxic granulation. Causes: Vacuoles may be a degenerative result of prolonged contact with the EDTA anticoagulant. However, when accompanied by toxic granulation and/or Döhle bodies, these vacuoles may be attributed to any of the conditions associated with toxic granulation: severe infectious processes or sepsis, severe inflammatory states, trauma, burns, certain cytokine medications, pregnancy, bone marrow response to myelosuppressive therapy, and uremia.
· See:
{kind=link}
{kind=link}
{kind=link}
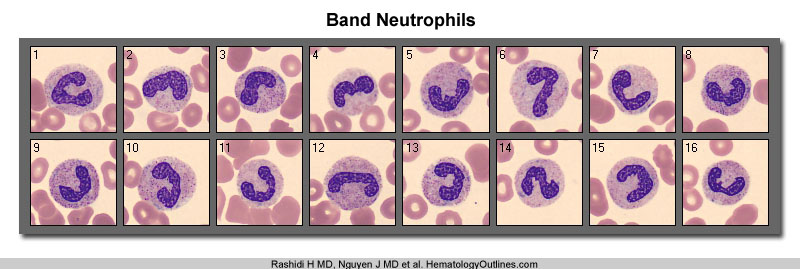
· Bands/ immature neutrophils: Immature neutrophils are normal in circulation in small numbers, but if there is a percentage increase of them, there is said to be a "left shift." This may happen when an acute infection stimulates increased neutrophil production, causing the bone marrow to prematurely release some white blood cells (WBCs) before they have matured to the neutrophil stage.
· A band cell (also called a band neutrophil or stab cell) is an immature white blood cell (WBC) released by the bone marrow into the blood. It is a cell undergoing granulopoiesis, derived from a metamyelocyte, and leading to a mature granulocyte. It is characterized by a curved nucleus (C or S-shaped), but not lobar. The term "band cell" implies a granulocytic lineage (e.g., neutrophils). A count of band neutrophils is used to measure inflammation. In adults, the reference range for neutrophil band cells is 3 to 5% of white blood cells (WBCs), or up to 0.7 x 10^9/L. An excess of bands is called bandemia. It is a signifier of infection (or sepsis) or inflammation. Measuring it can inform the approach to appendicitis.
· Other immature forms that may sometimes be seen on a blood smear include myelocyte, metamyelocyte, or even promyelocyte and myeloblast.
· See:
{kind=link}
{kind=link}
· http://classconnection.s3.amazonaws.com/913/flashcards/1300913/jpg/band_neutrophil21334523938623.jpg
{kind=link}
{kind=link}
{kind=link}
· SIRS (Systemic Inflammatory Response Syndrome): SIRS is a serious condition related to systemic inflammation, organ dysfunction, and organ failure. It is caused by abnormal regulation of various cytokines. It is also closely related to sepsis, in which patients meet the SIRS criteria and have a suspected or proven infection. Manifestations of SIRS in adults include, but are not limited to:
· Body temperature < 36°C(96.8°F) or > 38°C(100.4°F).
· Heart rate > 90 bpm
· Tachypnea (high respiratory rate), with > 20 breath/min; or an arterial partial pressure of CO2 < 4.3 kPa (32 mmHg).
· WBCs < 4000 cells/mm³ (4 x 109 cells/L) or > 12,000 cells/mm³ (12 x 109 cells/L); or the presence of > 10% immature neutrophils (band forms). Band forms greater than 3% are called bandemia or a "left shift."
· When SIRS is due to an infection, it is considered sepsis.
· Non-infectious causes of SIRS include trauma, burns, pancreatitis, ischemia, and hemorrhage. Other causes include adrenal insufficiency (Addison’s disease), pulmonary embolism, cardiac tamponade, complicated aortic aneurysm, a complication of surgery, drug overdose, anaphylaxis, etc.
Associated with hematological malignancies
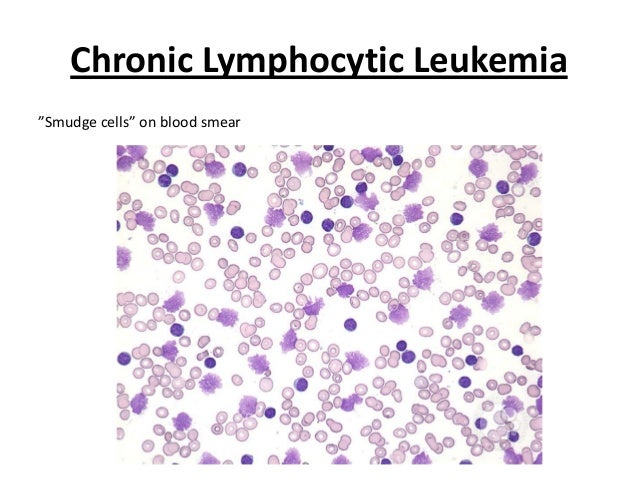
· Smudge cells (also sometimes called basket cells) are leukocytes that have been damaged during the preparation of the peripheral blood smear. This usually occurs due to cellular fragility. They are typically seen in chronic lymphocytic leukemia (CLL). However, they may be an artifact of slide preparation.
· A study concluded that the percentage of smudge cells on a blood smear is readily available and an independent factor predicting overall survival in CLL (Reference: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2668708/ )
· See:
{kind=link}
{kind=link}
{kind=link}
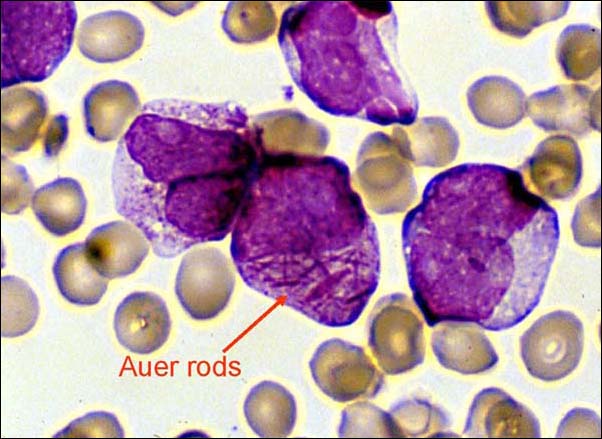
· Auer rods: elongated, bluish-red rods composed of fused lysosomal granules, seen in the cytoplasm of myeloblasts, promyelocytes, and monoblasts and in patients with acute myelogenous leukemia (AML).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Hairy cells are characterized by their fine, irregular pseudopods and immature nuclear features. Hairy cell leukemia (also called leukemic reticuloendotheliosis) is a rare malignant disorder that usually occurs in middle-aged patients over fifty. The first symptoms of the disease include weakness and lethargy. An enlarged spleen occurs in 80% of patients. Bone marrow aspiration is often unsuccessful because of complete infiltration by hairy cells, resulting in a dispersed spongy web of cells in an increased meshwork of reticulin fiber.
· See:
· http://www.ijpmonline.org/articles/2012/55/1/images/IndianJPatholMicrobiol_2012_55_1_61_94858_f1.jpg
{kind=link}
{kind=link}
· Plasma cells: A plasma cell is a mature B lymphocyte that is specialized for immunoglobulin production. Plasma cells are rarely found in the peripheral blood. They comprise from 0.2% to 2.8% of the bone marrow white cell count. Mature plasma cells are often oval or fan-shaped, measuring 8 – 15 µm. The nucleus is eccentric and oval. The nucleus-to-cytoplasm ratio typically ranges from 2:1 to 1:1. The nucleus may be bilobed or multilobed, especially in patients with lymphoid blood dyscrasias. The perinuclear zone is very distinct, appearing white in the profoundly basophilic cytoplasm. The nuclear chromatin is condensed and highly patchy, appearing as dark blocks on a reddish-purple background. The cytoplasm stains deep blue to gray-blue, depending on the stain and the ribosomal content of the individual cell. Plasma cells are seen in multiple myeloma, plasma cell leukemia, Waldenstrom's macroglobulinemia, and MGUS (monoclonal gammopathy of uncertain significance).
· The morphological characteristics of clonal plasma cells span the same morphological spectrum found in other plasma cell dyscrasias, where mature plasma cells are oval with abundant basophilic cytoplasm, and the nucleus is round and eccentrically located with a marked perinuclear hof. Immature plasma cells have dispersed nuclear chromatin, prominent nucleoli, and a high nuclear-to-cytoplasmic ratio. However, at times, clonal plasma cells in patients with plasma cell leukemia (PCL) may be indistinguishable from myeloblasts. Immunophenotypically, these clonal plasma cells express either κ or λ light chains, but not both, and demonstrate increased expression of CD138, CD38, and CD20 (especially pPCL).
· Cells rich in carbohydrate-containing IgA may have flame–pink cytoplasm and are called flame cells.
· See:
{kind=link}
{kind=link}
· Flame cell:
{kind=link}
{kind=link}
Leukemias
· Αcute lymphoblastic leukemia (ALL; also known as acute lymphocytic leukemia and acute lymphoid leukemia): usually strikes children between the ages of 2 to 10. The second peak in incidence is seen in elderly patients. Only half of patients with ALL have elevated leukocytes, and some may not have lymphoblasts in their peripheral blood. Neutropenia, thrombocytopenia, and anemia are usually present. Patients present with fatigue, fever, and bleeding. There is often lymph node enlargement. Enlargement of the spleen and of the liver may be seen.
· See:
{kind=link}
{kind=link}
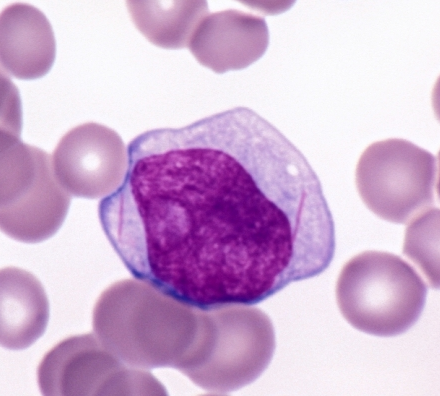
· Acute myeloid leukemia (AML, also known as acute myelogenous leukemia) is the most common leukemia in children less than 1 year of age. It is rare in older children and adolescents, but the second peak of incidence occurs among adults aged 40 years. The patient usually has an elevated white blood cell count, and myeloblasts are present. Anemia, thrombocytopenia, and neutropenia give rise to clinical findings such as pallor, bruising and bleeding, fever with infections, and fatigue. Bone and joint pain are the first symptoms in 25% of patients. An enlarged spleen is seen in 50% of all AML patients, but lymph node enlargement is rare. Auer rods: elongated, bluish-red rods composed of fused lysosomal granules, seen in patients with acute myelogenous leukemia (AML).
· See:
{kind=link}
{kind=link}
{kind=link}
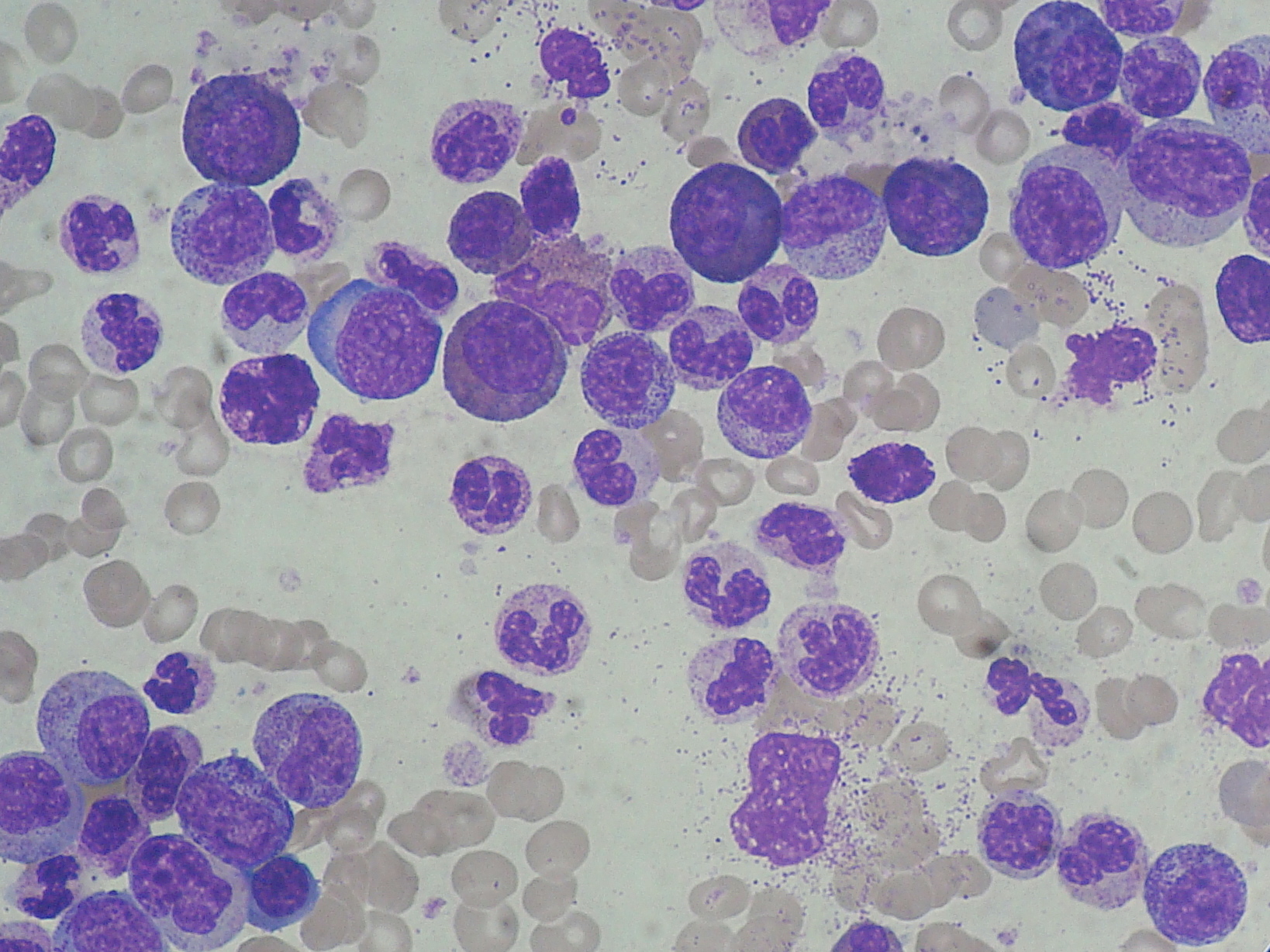
· Chronic myelogenous leukemia (CML; also called chronic myeloid, myelocytic, or myelogenic leukemia and chronic granulocytic leukemia (CGL) can occur at any age but is most common after the age of 45 years. Weight loss and fatigue are often the initial symptoms. There is usually massive spleen enlargement, which may cause left upper abdominal pain. There is anemia, markedly elevated leukocyte counts, thrombocytosis, eosinophilia, basophilia, and a predominance of myelocytes in the peripheral blood. Myeloblasts constitute fewer than 10% of circulating leukocytes. Occasional nucleated red blood cells are seen.
· See:
{kind=link}
{kind=link}
{kind=link}
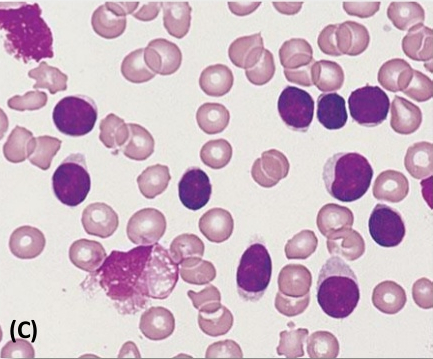
· Chronic lymphocytic leukemia (CLL, also known as chronic lymphoid leukemia) is the most common type of leukemia and usually occurs in older patients; it is rare in patients less than 40 years of age. The disease is generally discovered when other medical problems are present. Weakness, fatigue, and weight loss are usually seen. The malignant cell in CLL is generally a small, mature-appearing lymphocyte. Smudge cells are leukocytes that have been damaged during the preparation of the peripheral blood smear. This usually occurs due to cellular fragility. They are typically seen in chronic lymphocytic leukemia (CLL).
· See:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(Smudge cells)
· Hairy cell leukemia (HCL; also called leukemic reticuloendotheliosis): a rare malignant disorder that usually occurs in middle-aged patients over 50. The first symptoms of the disease include weakness and lethargy. An enlarged spleen occurs in 80% of patients. Bone marrow aspiration is often unsuccessful because of complete infiltration by hairy cells, resulting in a dispersed spongy web of cells in an increased meshwork of reticulin fiber. Hairy cells are characterized by their fine, irregular pseudopods and immature nuclear features.
· See:
· http://www.ijpmonline.org/articles/2012/55/1/images/IndianJPatholMicrobiol_2012_55_1_61_94858_f1.jpg
![http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/hairy_cell_leukemia_classic/hairy-cell-leukemia-[7-bl089-3].jpeg?Width=600&Height=450&Format=4](http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/hairy_cell_leukemia_classic/hairy-cell-leukemia-%5b7-bl089-3%5d.jpeg?Width=600&Height=450&Format=4){kind=link}
![http://www.pathpedia.com/education/eatlas/histopathology/bone_marrow/hairy_cell_leukemia_bone_marrow/hairy-cell-leukemia-[9-bm083-9].jpeg?Width=600&Height=450&Format=4](http://www.pathpedia.com/education/eatlas/histopathology/bone_marrow/hairy_cell_leukemia_bone_marrow/hairy-cell-leukemia-%5b9-bm083-9%5d.jpeg?Width=600&Height=450&Format=4){kind=link}
{kind=link}
· Hairy cell leukemia-variant (HCL – V):
{kind=link}
{kind=link}
{kind=link}
Platelets (Plts)
· Clamping of platelets and falsely low platelet count (pseudo-thrombocytopenia): Often, abnormal results will prompt additional testing. Under certain conditions, platelets may clump together and appear falsely low in number and/or large, so a blood smear may be performed to examine platelets directly under a microscope. A direct examination of a blood smear may be necessary to determine whether the platelets are genuinely giant or have clumped together during testing. If platelets are clumping, repeat testing may be performed using a different collection tube with an anticoagulant that prevents or minimizes platelet clumping.
· A pseudothrombocytopenia false-positive result may occur when automated platelet counting devices are used. The patient's blood sample is often examined under a microscope. If the clumping is visible and the platelet count appears normal, pseudothrombocytopenia may be concluded. A second sample run with a different anticoagulant, such as citrate (blue-top tube), to confirm pseudothrombocytopenia may be requested if there are doubts or concerns.
· A study concluded that in routine hematological practice, CPT (with trisodium citrate 17 mmol/l, pyridoxal 5'-phosphate 11.3 mmol/l, and Tris 24.76 mmol/l) can be an alternative anticoagulant to K3. EDTA. It is most suitable for automated complete blood count and useful in avoiding EDTA-induced platelet clumping (Reference: http://www.ncbi.nlm.nih.gov/pubmed/2110927 )
· Giant platelets: are platelets that are larger than 6.5 µm, or 75 to 100% the size of a normal red blood cell. MPV (mean platelet volume) is increased. A normal red cell is 6 – 8 µm in diameter. Normal platelets are approximately 1 – 4 µm, while large platelets are about 4 – 6.5 µm. Giant platelet is a term used to describe abnormally large platelets, i.e., as large as a normal red blood cell.
· Giant platelet disorders are rare, featuring abnormally large platelets, thrombocytopenia, and bleeding tendency.
· These may be seen in specific disorders such as immune thrombocytopenic purpura (ITP) or in rare inherited disorders such as Bernard-Soulier disease, gray platelet syndrome, and May–Hegglin anomaly.
· Platelet satellitism means platelets encircling a neutrophil and occurs when a patient has a serum factor that reacts to the anticoagulant EDTA.
Parasites
· See above: babesia & malaria.
Bacteria
· The primary function of the neutrophil is to stop or retard the action of foreign matter or infectious agents. The neutrophil accomplishes this by moving to the area of inflammation or infection, phagocytizing the foreign material, and killing and digesting the bacteria.
· Bacterial sepsis can result in a leukemoid reaction involving white cell counts of 100 000 X 106/L, the presence of myelocytes, and the appearance of toxic granulation.
· See:
{kind=link}
{kind=link}
(description on http://www.cdc.gov/relapsing-fever/clinicians/)
Fungi/ Yeasts
· The primary function of the neutrophil is to stop or retard the action of foreign matter or infectious agents. The neutrophil accomplishes this by moving to the area of inflammation or infection, phagocytizing the yeast, digesting and killing it.
· See:
{kind=link}
{kind=link}
INFECTIONS WITH HEMATOLOGICAL MANIFESTATIONS
· Leishmaniasis: Leishmaniasis is a disease caused by protozoan parasites of the genus Leishmania and spread by the bite of certain types of sandflies. The infection may occur in many other animals, including dogs & rodents. The disease can present in three main forms: cutaneous (skin), mucocutaneous, or visceral.
· The cutaneous form presents with skin ulcers, while the mucocutaneous form presents with ulcers of the skin, mouth, and nose, and the visceral form starts with skin ulcers and then later presents with fever, low red blood cells, and an enlarged spleen and liver.
· Signs & symptoms:
· There is an infection of the liver, spleen, and bone marrow, causing the classical features of:
· Night sweats, weakness, and anorexia are typical.
· Fever.
· Weight loss.
· Hepatomegaly (can be marked).
· Splenomegaly (often enormous).
· Anemia and pancytopenia (can lead to death from hemorrhage or infection).
· Hypergammaglobulinemia.
· Dark pigmentation of the skin is uncommon, but the name kala-azar is Hindi for black fever.
· Diagnosis:
· Leishmaniasis is diagnosed in the hematology lab by direct visualization of the amastigotes (Leishman-Donovan bodies). Buffy–coat preparations of peripheral blood or aspirates from marrow, spleen, lymph nodes, or skin lesions should be spread on a slide to make a thin smear and stained with Leishman stain or Giemsa stain for 20 minutes. Amastigotes are seen within monocytes or, less commonly, in neutrophils of peripheral blood and in macrophages of aspirates. Other indirect immunological diagnostic methods have been developed, including enzyme-linked immunosorbent assay, antigen-coated dipsticks, and direct agglutination test. Although these tests are readily available, they are not standard diagnostic tests because of their insufficient sensitivity and specificity. Several PCR tests are available for detecting Leishmania DNA, offering high specificity and sensitivity.
· Treatment: liposomal Amphotericin B (for the Visceral form in India, South America, and the Mediterranean, this is now the recommended treatment), antimony compounds (Sodium stibogluconate), miltefosine, paromomycin, fluconazole, and pentamidine.
· Typhoid fever (enteric fever):
· Typhoid fever is a potentially fatal multisystemic illness caused primarily by Salmonella enterica, subspecies enterica serovar typhi, and, to a lesser extent, related serovars paratyphi A, B, and C. The clinical syndromes associated with S. typhi and paratyphi are indistinguishable.
· Presentation: typhoid fever begins 7-14 days after ingestion of the organism. The fever pattern is stepwise, characterized by a daily rise in temperature that drops by the following morning. The peaks and troughs rise progressively over time.
· Over the course of the first week of illness, the gastrointestinal manifestations of the disease develop. These include diffuse abdominal pain and tenderness and, in some cases, fierce, colicky right upper quadrant pain. Monocytic infiltration inflames Peyer patches and narrows the bowel lumen, causing constipation that lasts throughout the illness. The individual then develops a dry cough, dull frontal headache, delirium, and an increasingly stuporous malaise. At approximately the end of the first week of illness, the fever plateaus at 103-104°F (39-40°C). The patient develops rose spots, which are salmon-colored, blanching, truncal, maculopapular, usually 1 –4 cm wide and fewer than five in number; these generally resolve within 2-5 days.
· During the second week of illness, the signs and symptoms listed above progress. The abdomen becomes distended, and soft splenomegaly is common. Relative bradycardia and a dicrotic pulse (a double beat, with the second beat weaker than the first) may develop.
· In the third week, the still febrile individual grows more toxic and anorexic with significant weight loss. The conjunctivae are infected, and the patient is tachypneic with a thready pulse and crackles over the lung bases. Abdominal distension is severe. Some patients experience foul, green-yellow, liquid diarrhea (pea soup diarrhea). The individual may descend into a typhoid state characterized by apathy, confusion, and even psychosis. Necrotic Peyer patches may cause bowel perforation and peritonitis. This complication is often unheralded and may be masked by corticosteroids. At this point, overwhelming toxemia, myocarditis, or intestinal hemorrhage may cause death.
· Some survivors become asymptomatic S typhi carriers.
· Lab features:
· Most patients with typhoid fever are moderately anemic and have an elevated ESR erythrocyte sedimentation rate), thrombocytopenia, and relative lymphopenia.
· Most also have a slightly elevated prothrombin time (PT), activated partial thromboplastin time (aPTT), and decreased fibrinogen levels.
· Circulating fibrin degradation products commonly rise to levels seen in subclinical disseminated intravascular coagulation (DIC).
· Liver transaminase and serum bilirubin values usually rise to twice the reference range.
· Mild hyponatremia and hypokalemia are common.
· A serum alanine aminotransferase (ALT)–to–lactate dehydrogenase (LDH) ratio of more than 9:1 appears to be helpful in distinguishing typhoid from viral hepatitis. A ratio greater than 9:1 supports a diagnosis of acute viral hepatitis, while a ratio of less than 9:1 supports typhoid hepatitis.
· Diagnosis: The criterion standard for diagnosis of typhoid fever has long been culture isolation of the organism. Cultures are widely considered 100% specific.
· Culture of bone marrow aspirate is 90% sensitive until at least 5 days after commencement of antibiotics. However, this technique is extremely painful, potentially outweighing its benefits.
· Blood, intestinal secretions (vomitus or duodenal aspirate), and stool culture results are positive for Salmonella typhi in approximately 85% – 90% of patients with typhoid fever who present within the first week of onset. They decline to 20% – 30% later in the disease course.
· Assays that identify Salmonella antibodies or antigens support the diagnosis of typhoid fever, but these results should be confirmed with cultures or DNA (PCR) evidence.
· The Widal test was the mainstay of typhoid fever diagnosis for decades. It is used to measure agglutinating antibodies against the H and O antigens of S typhi. Neither sensitive nor specific, the Widal test is no longer an acceptable clinical method.
· Indirect hemagglutination, indirect fluorescent Vi antibody, and indirect enzyme-linked immunosorbent assay (ELISA) for immunoglobulin M (IgM) and IgG antibodies to S typhi polysaccharide, as well as monoclonal antibodies against S typhi flagellin, are promising, but the success rates of these assays vary in the literature.
· Polymerase chain reaction (PCR) has been used for the diagnosis of typhoid fever with varying success.
· Treatment: Definitive treatment of typhoid fever is based on (antibiotic) susceptibility. As a general principle of antimicrobial treatment, intermediate susceptibility should be regarded as equivalent to resistance. Between 1999 and 2006, 13% of S typhi isolates collected in the United States were multidrug-resistant.
· Until susceptibilities are determined, antibiotics should be empiric, for which there are various recommendations. The authors of the Medscape article recommend combination treatment with ceftriaxone and ciprofloxacin when neither the sensitivities nor the geographical origin of the bacteria is known (Reference (Retrieved: December 28, 2015): http://emedicine.medscape.com/article/231135-medication#1 ).
· Prompt administration of high-dose dexamethasone reduces mortality in patients with severe typhoid fever without increased incidence of complications, carrier states, or relapse among survivors.
· Brucellosis:
· Brucellosis is a highly contagious zoonosis (animal disease) caused by ingestion of unpasteurized milk or undercooked meat from infected animals or close contact with their secretions. Brucella species are small, Gram-negative, nonmotile, non-spore-forming, rod-shaped (coccobacilli) bacteria that cause chronic disease.
· Four species infect humans: B. melitensis, B. abortus, B. suis, and B. canis. B. melitensis is the most virulent and invasive species; it usually affects goats and occasionally sheep. B. abortus is less virulent and is primarily a disease of cattle. B. suis is of intermediate virulence and chiefly infects pigs. B. canis affects dogs.
· Signs & symptoms: muscular pain and sweating are characteristic. The duration of the disease can vary from a few weeks to many months or even years.
· In the first stage of the illness, septicemia occurs, leading to the classic triad of undulant fevers, sweating (often with a characteristic smell likened to wet hay), and migratory arthralgia & myalgia.
· Lab features:
· Blood tests characteristically reveal leukopenia & anemia.
· There is also some elevation of the liver function tests, AST, and ALT.
· Diagnosis:
· Definite diagnosis of brucellosis requires the isolation of the organism from the blood, body fluids, or tissues, but serological methods may be the only tests available in many settings. Positive blood culture yield ranges between 40% and 70% and is less commonly positive for B. abortus than Β. melitensis or B. suis. Identification of specific antibodies against bacterial LPS and other antigens can be detected by the standard agglutination test (SAT), rose Bengal, 2-mercaptoethanol (2-ME), antihuman globulin (Coombs’), and indirect enzyme-linked immunosorbent assay (ELISA). SAT is the most used serology in endemic areas. However, due to the similarity of the O polysaccharide of Brucella to that of many other Gram-negative bacteria, the appearance of cross-reactions of class M immunoglobulins may occur.
· ELISA typically uses cytoplasmic proteins as antigens. It measures IgM, IgG, and IgA with better sensitivity and specificity than the SAT in the most recent comparative studies.
· Many varieties of PCR have been developed and found to have superior specificity and sensitivity in detecting both primary infection and relapse after treatment. However, some centers have reported persistent PCR positivity after clinically successful treatment, fueling the controversy about the existence of prolonged chronic brucellosis.
· Treatment: antibiotics such as tetracyclines, rifampicin, streptomycin, or gentamycin are effective against Brucella bacteria. However, using more than one antibiotic is necessary for several weeks because the bacteria reside within cells (intracellular).
· Pertussis (whooping cough):
· Pertussis, also known as whooping cough, is a highly contagious bacterial disease. Pertussis is caused by the bacterium Bordetella pertussis. It is an airborne disease that spreads quickly through the coughs and sneezes of an infected person. People are infectious to others from the start of symptoms until about three weeks into the coughing fits.
· Signs & symptoms: Initially, symptoms are usually like those of the common cold with a runny nose, fever & mild cough. This is then followed by weeks of severe coughing fits. Following a fit of coughing, a high-pitched whooping sound or a gasp may occur as the person inhales. The coughing may last for 10 or more weeks, hence the phrase "100-day cough. A person may cough so hard that they vomit, break ribs, or become very tired. Children less than one year old may have little or no cough and instead have apnea. The period between infection and the onset of symptoms is usually seven to ten days.
· Disease may occur in those who have been vaccinated, but symptoms are typically milder.
· Lab features:
· FBC (CBC): Although not specific, lymphocytosis is a diagnostic clue for pertussis.
· Diagnosis: Methods used in laboratory diagnosis include culturing of nasopharyngeal swabs on a nutrient medium, PRC, direct fluorescent antibody (DFA), and serological tests, including the complement fixation test. The bacteria can be recovered from the person only during the first three weeks of illness, rendering culturing and DFA useless after this period, although PCR may have some limited usefulness for an additional three weeks.
· Treatment: The macrolide antibiotics erythromycin, clarithromycin, or azithromycin are typically the recommended treatment.
· Prevention: by vaccination with the multicomponent acellular pertussis vaccine. Immunization does not confer lifelong immunity. A 2011 CDC study indicated that protection may last only 3 to 6 years. This covers childhood, the period of greatest exposure and highest risk of pertussis-related death.
· Granulocytes: are only PMNs (polymorphonuclear neutrophils), eosinophils, and basophils.
· Monocytes are not granulocytes (although they may contain fine granules).
· Lymphocytes don’t have granules. However, LGL (large cell granulocytes) have granules.
· Azurophil granules: e.g., on PMN’s cytoplasm are burgundy or merlot colored.
· Agranulocytosis: leucopenia (especially neutrophils). Granulocytes (neutrophils, basophils, eosinophils) drop to <500 cells/mm3. In neutropenia, the absolute neutrophil count is < 500 cells/mm3, and in agranulocytosis, it is <100 cells/mm3.
· Causes of agranulocytosis: drugs (antiepileptics, antithyroid (carbimazole, methimazole, propylthiouracil), antibiotics (penicillin, chloramphenicol, co-trimoxazole (sulfamethoxazole & trimethoprim)), gold, cytotoxic drugs, NSAIDs (non–steroidal anti-inflammatory drugs), allopurinol, mebendazole (to treat worms), antidepressants such as mirtazapine, antipsychotics such as the atypical antipsychotic clozapine, etc.
· Agranulocytosis – differential diagnosis: aplastic anemia, MDS (myelodysplasia), leukemia, paroxysmal nocturnal hemoglobinuria. Bone marrow examination.
· Neutrophilia: high number of neutrophil granulocytes in peripheral blood.
· Causes of neutrophilia: bacterial infections (especially pyogenic), tissue destruction (surgical or other trauma, crush injury, burns, myocardial infarction, or another infarct such as pulmonary infarction), inflammatory disorders (e.g., vasculitis). Acute hemorrhage, acute hemolysis, metabolic disorders (acidosis, uremia, gout, toxins), hematological malignancies (e.g., chronic myeloproliferative diseases such as CML (chronic myelogenous leukemia), Hodgkin’s lymphoma), solid tumors, drugs [including corticosteroids, epinephrine (adrenaline), antibiotics such as tetracycline, lithium, and others], nervousness (may cause slight increase), splenectomy, appendicitis, pancreatitis, and physiological during pregnancy, exercise and surgical procedures.
· GM – CSF (Granulocyte-macrophage colony-stimulating factor, also known as colony-stimulating factor 2 (CSF2)) is used as a medication to stimulate the production of white blood cells and thus prevent neutropenia following chemotherapy. It stimulates the proliferation of precursors to neutrophils, eosinophils, and monocytes/macrophages.
· Note: a study concluded that there is a systemic increase in polymorphonuclear (PMN) activity and a decrease in T-lymphocyte activity in patients with acute coronary syndrome. This contrasts with the pattern of cellular activation observed at sites of local inflammation within atherosclerotic plaques, suggesting that two independent inflammatory processes (local and systemic) may be involved in the pathogenesis of this syndrome (Reference: http://www.ncbi.nlm.nih.gov/pubmed/9430368).
· Shift to the left: a premature release of immature cells of the neutrophil series (bands, metamyelocytes, myelocytes, promyelocytes, and rarely myeloblasts) in peripheral blood. A severe neutrophilia with a left shift is referred to as a leukemoid reaction (see below). The leukocyte alkaline phosphatase (LAP) score (the amount of alkaline phosphate per neutrophil) will increase. LAP (Leukocyte alkaline phosphatase) score is low in CML (chronic myelogenous leukemia) but normal or increased in leukemoid reaction.
· In a severe infection, a peripheral blood smear may show toxic granulation in the neutrophils.
· Neutropenia: absolute neutrophil count in peripheral blood < 2 000/cmm. Grade: mild (neutrophils 1,000 – 1,500/cmm; no increased risk for infection), moderate (neutrophils 500 – 1,000/cmm; mild risk for infection), severe (neutrophils < 500/cmm; significant risk for infection).
· Causes of neutropenia: infections (overwhelming bacterial infections, septicaemia, miliary TB (tuberculosis), HIV, influenza, infectious mononucleosis etc.), viral infections (mild neutropenia), morning pseudoneutropenia (transient reduction in the measured neutrophil count from peripheral samples; it is noticed in some patients who are taking antipsychotic medication such as clozapine and risperidone), drugs [such as antipsychotics (e.g. chlorpromazine, clozapine), ticlopidine (an antiplatelet), antimicrobials (e.g. sulfonamides e.g. sulfamethoxazole + trimethoprim; chloramphenicol), analgesics (phenylbutazone, oxyphenylbutazone), phenytoin (antiepileptic and also antiarrhythmic), flecainide (antiarrhythmic), indomethacin (a NSAID non-steroidal anti-inflammatory drug), antithyroid drugs (for hyperthyroidism e.g. carbimazole, methimazole, and propylthiouracil (PTU); the last may also cause fulminant liver failure), cytotoxic drugs, chemotherapy (for cancer or for autoimmune disorders), autoimmune neutropenia [including Felty’s syndrome (rheumatoid arthritis, splenomegaly and neutropenia), collagen diseases [including SLE (systemic lupus erythematosus), neonatal isoimmune neutropenia, drug induced], ineffective hematopoiesis (megaloblastic anemia; vitamin B12 and/or folate deficiency), abnormal pooling (hypersplenism), bone marrow replacement (leukemia, chronic myeloproliferative disorders, myelodysplastic syndromes, myeloma, lymphoma), bone marrow hypoplasia (aplastic anemia, arsenic poisoning), other rare conditions (cyclic neutropenia, congenital neutropenia, chronic idiopathic neutropenia, Kostman’s syndrome (severe congenital neutropenia) etc.), radiation exposure, copper deficiency, hemodialysis etc.
SYNDROMES ASSOCIATED WITH NEUTROPENIA & RECURRENT INFECTIONS/IMMUNODEFICIENCY-RELATED SYNDROMES
· Felty's syndrome: a combination of RA (rheumatoid arthritis), splenomegaly, and neutropenia. The condition is more common in those aged 50 – 70 years and is more prevalent in females than males and more in Caucasians than blacks. It is a deforming but inactive disease and seropositive for RF (rheumatoid factor).
· Chediak–Higashi syndrome: a rare autosomal recessive disorder that arises from a mutation of a lysosomal trafficking regulator protein, which leads to a decrease in phagocytosis that results in recurrent pyogenic infections, partial ocular-cutaneous albinism, and peripheral neuropathy.
· People with CHS have light skin and silvery hair (partial albinism) and frequently complain of solar sensitivity and photophobia.
· Neutropenia is common, leading to frequent infections, especially with Staphylococcus aureus and Streptococci. Also, neuropathy is common. The infections involve mucous membranes, skin, and the respiratory tract. Affected children are susceptible to infections.
· It is also associated with periodontal disease of the deciduous dentition and bleeding disorders.
· The diagnosis is confirmed by bone marrow smears that show "giant inclusion bodies" in the cells that develop into white blood cells (leukocyte precursor cells). CHS can be diagnosed prenatally by examining hair from a fetal scalp biopsy or by testing leukocytes from a fetal blood sample.
· http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/chediak-higashi_syndrome/chediak-higashi-syndrome-[3-bl050-3].jpeg?Width=600&Height=450&Format=4http://image.slidesharecdn.com/011209-jlevine-myeloidcelldisorders-110727140520-phpapp01/95/011209-myeloid-cell-disorders-33-728.jpg?cb=1311775763
· Wiskott–Aldrich syndrome (WAS; also called eczema – thrombocytopenia –immunodeficiency syndrome): a rare Χ – linked recessive disease characterized by eczema, thrombocytopenia, immune deficiency, and bloody diarrhea (due to low platelets). Due to its mode of inheritance, most patients are male. The first signs of WAS are usually petechiae and bruising resulting from a low platelet count. Spontaneous epistaxis and bloody diarrhea are common.
· Eczema develops within the first month of life, and recurrent bacterial infections develop for three months. Splenomegaly is not uncommon. Most WAS children develop at least one autoimmune disorder, and cancers (mainly leukemia and lymphoma) develop in up to 1/3 of patients. Immunoglobulin M (IgM) levels are reduced, IgA and IgE are elevated, and IgG levels can be normal, decreased, or elevated.
{kind=link}
{kind=link}
HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS (HLH)
· Primary hemophagocytic lymphohistiocytosis (i.e., familial erythrophagocytic lymphohistiocytosis [FEL]), an inherited form of hemophagocytic lymphohistiocytosis syndrome, is a heterogeneous autosomal recessive disorder.
· Secondary hemophagocytic lymphohistiocytosis (i.e., acquired hemophagocytic lymphohistiocytosis) occurs after strong immunologic activation, such as that which can occur with systemic infection, immunodeficiency, or underlying malignancy.
· Both forms are characterized by the overwhelming activation of normal T lymphocytes and macrophages, invariably leading to clinical and hematologic alterations and death in the absence of treatment.
· Incidence is reported to be 1.2 cases per million persons per year. However, unpublished observations estimate one case per every 50,000 births.
· The age of onset is usually in people younger than 1 year for the familial form, but can be later for the sporadic secondary form, usually after age 6 years; however, familial forms have been reported in individuals as old as 8 years, and adult-onset has been reported. Thus, no age criteria have been established, and there is no upper age limit. The onset of HLH occurs under the age of 1 year in about 70% of cases. Familial HLH should be suspected if siblings are diagnosed with HLH or if symptoms recur when therapy has been stopped. Each full sibling of a child with familial HLH has a 25% chance of developing the disease, a 50% chance of carrying the defective gene (which is very rarely associated with any risk of disease), and a 25% chance of not being affected and not carrying the gene defect.
· Clinical signs: HLH clinically manifests with fever, hepatosplenomegaly, lymphadenopathy, jaundice, and a rash.
· The current (2008) diagnostic criteria for HLH are:
· a) A molecular diagnosis consistent with HLH. These include the identification of pathologic mutations of PRF1, UNC13D, or STX11
· ΟR
· b) Fulfillment of five out of the eight criteria of the Histiocyte Society below:
· 1) Fever (seven or more days of a temperature as high as 38.5°C (101.3°F))
· 2) Splenomegaly (a palpable spleen greater than 3 cm below the costal margin)
· 3) Cytopenias affecting at least two of three lineages in the peripheral blood:
· Hemoglobin <9 g/100 ml (in infants <4 weeks: haemoglobin <10 g/100 ml)
· Platelets <100×109/L
· Absolute neutrophils less than 1000/µ
· 4) Hypertriglyceridemia (fasting triglycerides greater than or equal to 265 mg/100 ml or greater than 2 mmol/L or levels greater than 3 standard deviations above the age-adjusted reference range value) and/or hypofibrinogenemia (fibrinogen less than 1.5 g/L or levels greater than 3 standard deviations below the age-adjusted reference range value)
· 5) Ferritin ≥ 500 ng/ml
· 6) Haemophagocytosis in the bone marrow, spleen, or lymph nodes without evidence of malignancy.
· 7) Low or absent natural killer (NK) activity. Because natural killer (NK) cell function or activity is decreased in as many as 90% of patients with hemophagocytic lymphohistiocytosis (HLH), it is one of the most useful laboratory tests. NK cell number is usually not diagnostic.
· 8) Soluble CD25 (soluble IL-2 receptor) >2400 U/ml (or per local reference laboratory)
·Also, in the case of familial HLH, no evidence of malignancy should be apparent.
· It should be noted that not all five out of eight criteria are required for the diagnosis of HLH in adults, and a high index of suspicion is required for diagnosis, as delays result in increased mortality. The diagnostic criteria were developed in pediatric populations and have not been validated for adult HLH patients.
· Attempts to improve the diagnosis of HLH have included the use of the H Score:
· Other signs & symptoms include:
· Rash: skin findings occur in more than 50% of patients, including scaly and waxy lesions and rashes on the scalp and behind the ear. As many as 65% of patients have a non-specific rash, often vaguely termed maculopapular, although it has also been described as ranging from erythroderma to generalized purpuric macules and papules, and to morbilliform eruptions.
· Swollen or hemorrhagic gums that can result in tooth loss; feeding problems (especially prominent in infants); abdominal pain, vomiting, diarrhea, and weight loss.
· Clinical findings also include evidence of infection due to decreased immunity and white cell killing defects, easy bruising, and pallor (related to pancytopenia secondary to bone marrow infiltration or splenic sequestration).
· One Swedish study mentions that about 75% of patients have some form of CNS involvement, with half showing neurologic symptoms, including seizures, ataxia, hemiplegia, mental status changes, or simply irritability.
· Lab features:
· Evidence of coagulopathy with an increased aPTT.
· Jaundice is often present due to hyperbilirubinemia.
· The blood count typically shows pancytopenia (anemia, neutropenia, and thrombocytopenia).
· The bone marrow may show hemophagocytosis.
· The liver function tests (AST & ALT) are usually elevated. Hypoalbuminemia is common.
· The serum CRP, ESR, and ferritin levels are markedly elevated. In pediatric populations, ferritin levels above 10,000 are highly sensitive and specific for the diagnosis of HLH. However, ferritin's diagnostic utility is lower in adult HLH patients. Ferritin has been observed to be a marker of hemophagocytic lymphohistiocytosis, with serum ferritin levels paralleling the course of the disease. A retrospective study evaluated the value of a ferritin level>500 ng/mL for diagnosing hemophagocytic lymphohistiocytosis in 344 consecutive patients admitted to the medical intensive care unit. The data suggested that a higher ferritin cutoff value may improve the utility of ferritin in the diagnosis of secondary hemophagocytic lymphohistiocytosis in the critical care setting.
· The serum fibrinogen level is usually low, and the D-dimer level is elevated.
· Sphingomyelinase is elevated.
· Causes: Epstein-Barr virus (EBV) infection is most commonly linked with hemophagocytic lymphohistiocytosis. HLH may be a complication of dengue fever, although this association is unusual. There is also an association with scrub typhus.
· Associated illnesses: infection, malignancy, rheumatologic, and immunodeficiency syndromes are common in patients with HLH. It is important to identify these conditions because effective treatment may lead to clinical improvement in HLH and allow the patient to avoid more toxic therapy (e.g., hematopoietic cell transplantation). However, evaluation for these associated syndromes should not delay diagnostic testing or initiation of HLH-specific treatment in those who are acutely ill.
· Infections: HLH is often associated with infections, especially viral. Common viruses include Epstein-Barr virus (EBV), cytomegalovirus (CMV), parvovirus, herpes simplex virus (HSV), varicella-zoster virus (VZV), measles, human herpesvirus-8 (HHV8; it causes Kaposi’s sarcoma), H1N1 influenza virus, parechovirus, and HIV, alone or in combination.
· Malignancy: HLH has been associated with malignancies, most commonly lymphoid cancers, including T, NK, and anaplastic large cell lymphomas, and leukemias. B-cell lymphoblastic leukemia, myeloid malignancies, and solid tumors occurring in association with HLH have also been noted. Rarely, the diagnosis of HLH may precede the identification of the malignancy. The survey of adult HLH patients reported that nearly 50% were associated with malignancies.
· Rheumatologic disorders/ macrophage activation syndrome) MAS): HLH can occur in the setting of rheumatologic disorders. The most common association is in children with systemic juvenile idiopathic arthritis (systemic JIA, formerly called Still's disease). The term macrophage activation syndrome (MAS) is used to describe a hemophagocytic syndrome that develops in children with JIA and other rheumatologic conditions. MAS should be thought of as HLH in the setting of a rheumatologic disorder rather than as a separate syndrome.
· Immunodeficiency: HLH has been found in patients with inherited immunodeficiency disorders, including those due to mutations associated with HLH and others. Acquired immunodeficiencies have also been associated with HLH, including HIV/AIDS, hematopoietic cell transplantation, or kidney or liver transplant.
· Workup:
· FBC, biochemistry (see above).
· The presence of a PRF1 gene mutation (for the inherited form) can be determined based on flow cytometry by staining perforin contained in lymphocytes.
· The finding of a distinctive Th1/Th2 cytokine pattern (significant increase of interferon and interleukin-10 with a slightly increased or normal level of IL-6) may represent a useful biomarker for early diagnosis, differential diagnosis, and disease monitoring.
· A skin biopsy can be performed for histological examination.
· Lymph node biopsy, bone marrow biopsy, or liver biopsy may demonstrate the characteristic hemophagocytosis.
{kind=link}
{kind=link}
{kind=link}
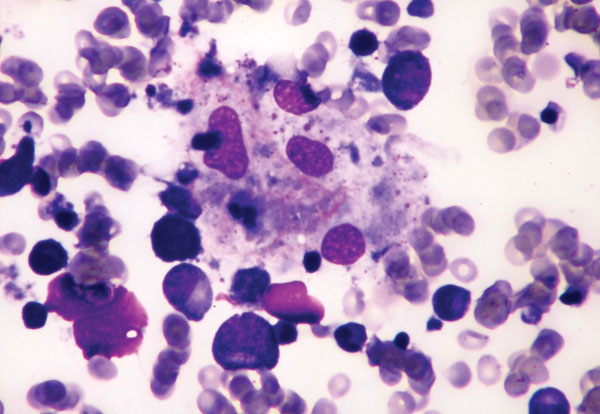{kind=link}
{kind=link}
· Treatment:
· Antineoplastic agents:
· Etoposide (VP-16; inhibits topoisomerase II and results in DNA strand breakage)
· Methotrexate (an antimetabolite that inhibits dihydrofolate reductase, hindering DNA synthesis and cell reproduction in malignant cells).
· Corticosteroids:
· Dexamethasone (decreases inflammation by suppressing migration of polymorphonuclear leukocytes and reducing capillary permeability).
· Immunosuppressants:
· Cyclosporine Cyclic polypeptide (suppresses some humoral immunity and, to a greater extent, cell-mediated immune reactions).
· Immune therapy:
· Immunoglobulins (IV): neutralize circulating myelin antibodies through anti-idiotypic antibodies; down-regulate proinflammatory cytokines, including IFN-gamma; block Fc receptors on macrophages; suppresses inducer T and B cells and augments suppressor T cells; blocks complement cascade; promotes remyelination, and may increase CSF IgG (10%)).
· anti-thymocyte globulin (ATG)
· Prognosis: the overall mortality is 50%. Poor prognostic factors include HLH associated with malignancy, with 50% of the patients dying by 1.4 months compared to 22.8 months for non-tumor-associated HLH patients. Secondary HLH in some individuals may be self-limited because patients can fully recover after receiving only supportive medical treatment (i.e., IV immunoglobulin alone). However, long-term remission without the use of cytotoxic and immunosuppressive therapies is unlikely in most adults with HLH and in those with CNS involvement.
No comments:
Post a Comment