
Dr. James Manos (MD)
January 5, 2016
Review: Tips in Medical Biochemistry
Volume (8)
CONTENTS
LIVER PROBLEMS
Jaundice – overview
Jaundice – diagnostic approach
Jaundice – table of diagnostic tests
Jaundice – diagnostic approach (algorithm)
Neonatal jaundice
Hyperbilirubinemia in the term newborn
Hyperbilirubinemia in the newborn – algorithm
Hyperbilirubinemia with abnormal liver function tests (LFTs) – diagnostic considerations
Child-Pugh score
Conjugated hyperbilirubinemia – overview
Conjugated hyperbilirubinemia –workup
Conjugated hyperbilirubinemia – causes
Unconjugated hyperbilirubinemia – causes
Hemolysis/ hemolytic anemia (overview)
Jaundice in the traveler to the tropics – differential diagnosis
LIVER PROBLEMS
· Jaundice – overview: jaundice (also known as icterus) is a yellowish pigmentation of the skin, the conjunctival membranes over the sclerae (whites of the eyes), and other mucous membranes caused by high blood bilirubin levels. This hyperbilirubinemia subsequently causes increased levels of bilirubin in the extracellular fluid. The concentration of bilirubin in blood plasma is normally below 1.2 mg/dl (<25 μmol/L). A concentration higher than approximately 3 mg/dL (>50 µmol/L) leads to jaundice. Jaundice may be prehepatic, hepatocellular (hepatic), or post-hepatic.
· Prehepatic jaundice: it is caused by anything which causes an increased rate of hemolysis, i.e., the breakdown of red blood cells (RBCs). Unconjugated bilirubin comes from the breakdown of the heme pigment found in red blood cells' hemoglobin. The increased breakdown of red blood cells leads to an increase in the amount of unconjugated bilirubin present in the blood and deposition of this unconjugated bilirubin into various tissues can lead to a jaundiced appearance. Kernicterus is associated with increased unconjugated bilirubin; neonates are especially vulnerable to this due to increased permeability of the blood-brain barrier. Unconjugated hyperbilirubinemia results from a derailment of the necessary bilirubin conjugation in the hepatocyte (liver cell). This problem may occur before bilirubin has entered the hepatocyte or within the liver cell. Excessive heme metabolism, from hemolysis or reabsorption of a large hematoma, results in significant increases in bilirubin, which may overwhelm the conjugation process and lead to a state of unconjugated hyperbilirubinemia.
· Hemolytic anemias usually result in mild bilirubin elevation, to about 5 mg/ dL (85.5 μmol/ L), with or without clinical jaundice.
· Causes of prehepatic jaundice: hemolysis. Hemolytic anemias result from abnormal red blood cell survival times. These anemias may occur because of membrane abnormalities (e.g., hereditary spherocytosis); enzyme abnormalities (e.g., glucose-6-phosphate dehydrogenase (G6PD) deficiency); autoimmune disorders, drugs, and defects in hemoglobin structure such as sickle cell disease and the thalassemias; hemolytic uremic syndrome (HUS); malaria, etc. Other causes include abnormalities in bilirubin metabolism such as Gilbert’s syndrome, and Crigler – Najjar syndrome.
· On the FBC/ CBC (full/ complete blood count) hemolysis is indicated by increased reticulocytes.
· Causes of hepatocellular (hepatic) jaundice: hepatitis; hepatotoxic drugs; infections such as leptospirosis; alcoholic liver disease; primary biliary cirrhosis, medications (such as acetaminophen (paracetamol) overdose), toxins [e.g., Amanita phalloides mushrooms, CCl4 (carbon tetrachloride), solvents such as toluene – e.g., in ‘’glue sniffing’’], etc.
· Jaundice seen in the newborn, known as neonatal jaundice, is common in newborns as liver machinery for the conjugation and excretion of bilirubin does not fully mature until approximately two weeks of age.
· In hepatic jaundice, there is invariably cholestasis.
· Causes of post-hepatic jaundice: gallstones, pancreatic cancer; liver flukes; biliary atresia; cholangiocarcinoma; pancreatitis; cholestasis of pregnancy; pancreatic pseudocysts, etc.
· Differential diagnosis: Jaundice can be caused by a malfunction in any of the three phases of bilirubin production.
· Pseudojaundice can occur with excessive ingestion of foods rich in beta-carotene (e.g., squash, melons, and carrots) called carotenemia. Unlike true jaundice, carotenemia does not result in scleral (whites of the eyes) icterus or elevation of the bilirubin level.
{kind=link}
· Jaundice – diagnostic approach: First-line serum testing in a patient presenting with jaundice should include a full/ complete blood count (FBC/ CBC) and determination of bilirubin (total and direct fractions), aspartate transaminase (AST; also known as SGOT), alanine transaminase (ALT; also known as SGPT), gamma-glutamyl transpeptidase (GGT), and alkaline phosphatase (ALP) levels.
· A CBC is useful in detecting hemolysis, which is indicated by the presence of fractured red blood cells (schistocytes) and increased reticulocytes on the smear.
· AST (SGOT) and ALT (SGPT) are markers of hepatocellular injury. They can be less helpful in patients with chronic liver disease, because levels can be normal or only slightly elevated when there is little liver parenchyma left to damage.
· Acute viral hepatitis may cause the levels of ALT (SGPT to rise several thousand units per liter.
· Levels greater than 10 000 U/ L usually occur in patients with acute injury to the liver from another source e.g., medications such as acetaminophen (paracetamol) or ischemia.
· Patients with acute alcoholic hepatitis have AST and ALT levels that rise to several hundred units per liter.
· With alcohol-induced damage, the ratio of AST (SGOT) to ALT (SGPT) is usually greater than 1, whereas infectious causes of hepatitis typically cause a greater elevation in ALT than in AST.
· Alkaline phosphatase (ALP) and gamma-glutamyltransferase (GGT) are markers for cholestasis. As bile obstruction progresses, the levels of these two markers rise several times above normal.
· The second-line serum investigations may include tests for hepatitis A IgM antibody, hepatitis B surface antigen and core antibody, hepatitis C antibody, and autoimmune markers such as antinuclear, smooth muscle, and liver-kidney microsomal antibodies. An elevated amylase level would corroborate the presence of pancreatitis.
· Jaundice – table of diagnostic tests:
· a) Prehepatic jaundice: total bilirubin: normal or increased; conjugated bilirubin: normal; unconjugated bilirubin: normal or increased; urobilinogen: normal or increased; urine color: normal; stool color: normal; ALP (alkaline phosphatase) levels: normal; ALT & AST levels: normal; conjugated bilirubin in urine: not present; splenomegaly may be present.
· Features of hemolysis: in hemolysis unconjugated bilirubin is increased (while conjugated bilirubin is normal); urinary urobilinogen is increased; urine color is normal; stool color is normal; ALP (alkaline phosphatase) levels are normal; AST (SGOT) & ALT (SGPT) levels are normal; conjugated bilirubin in urine is not present; splenomegaly may be present.
· On the FBC/ CBC (full/ complete blood count) hemolytic anemia is indicated by decreased hemoglobin and/or hematocrit (a sign of anemia) and is also indicated by the presence of increased reticulocytes that also are present on the peripheral blood smear (polychromatophilic erythrocytes).
· On hemolysis, there are also elevated levels of LDH lactate dehydrogenase, and decreased levels of plasma haptoglobin. Direct antiglobulin test (DAT; also called direct Coombs test) is positive on autoimmune hemolytic anemia.
· Other features of hemolytic anemia include increased free serum hemoglobin, hemoglobin in the urine (hemoglobinuria) and hemosiderin present in the urine (hemosiderinuria).
· Moreover, methemalbuminemia (the presence of methemalbumin in the circulating blood) is indicative of intravascular hemolysis.
· In autoimmune hemolytic anemia and hereditary spherocytosis, there is spherocytosis (presence of spherocytes) in the peripheral blood smear.
· Fractured red blood cells (schistocytes) in the blood smear are indicative of microangiopathic hemolytic anemia (MAHA) or other cause for intravascular hemolysis including disseminated intravascular coagulopathy (DIC)).
· Microangiopathic hemolytic anemia may occur in conditions such as hemolytic-uraemic syndrome (HUS), thrombotic thrombocytopenic purpura (TTP; Moschcowitz syndrome), DIC (disseminated intravascular coagulation), pro/eclampsia, malignant hypertension; or as the RBCs (red blood cells) pass across a damaged or prosthetic heart valve. Other causes include severe burns, uremia, toxins, MDS (myelodysplastic syndrome), march hemoglobinuria, and hemolytic anemias caused by physical agents.
· b) Hepatic jaundice: total bilirubin: increased; conjugated bilirubin: increased; unconjugated bilirubin: increased; urobilinogen: decreased; urine color: dark; stool color: normal or pale; ALP (alkaline phosphatase) levels: increased; ALT & AST levels: increased; conjugated bilirubin in urine: present; splenomegaly may be present.
· c) Post-hepatic jaundice: total bilirubin: increased; conjugated bilirubin: increased; unconjugated bilirubin: normal; urobilinogen: decreased or negative; urine color: dark (conjugated bilirubin); stool color: pale; ALP (alkaline phosphatase) levels: increased; ALT & AST levels: increased; conjugated bilirubin in urine: present; splenomegaly: absent.
· Jaundice – diagnostic approach (algorithm):
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Hemolytic anemia:
{kind=link}
· Neonatal jaundice: is a yellowing of the skin and other tissues of a newborn infant. A bilirubin level > 85 μmol/l (5 mg/dL) leads to a jaundiced appearance in neonates [whereas in adults a level of 34 μmol/l (2 mg/dL) is needed for this to occur]. It is often seen in infants around the second day after birth, lasting until day 8 in normal deliveries, or to around day 14 in premature births.
· Physiological jaundice: Physiologic jaundice is clinically obvious in 50% of neonates during the first 5 days of life. In physiologic jaundice, the peak total serum bilirubin level is 5 – 6 mg/dL (86 – 103 µmol/L), occurs at 48 – 120 hours (2nd – 5th day) of age, and does not exceed 17 – 18 mg/dL (291 – 308 µmol/L). Characteristics: bilirubin is < 20 mg/dl; resolves early. Physiological jaundice is a common condition in which most infants develop visible jaundice due to the elevation of unconjugated bilirubin concentration during their first week. This pattern of hyperbilirubinemia has been classified into two functionally distinct periods:
· a) Phase one:
· i) Term infants: jaundice lasts for about 10 days with a rapid rise of serum bilirubin up to 204 μmol/l (12 mg/dL).
· ii) Preterm infants: jaundice lasts for about two weeks, with a rapid rise of serum bilirubin up to 255 μmol/l (15 mg/dL).
· b) Phase two: bilirubin levels decline to about 34 μmol/l (2 mg/dL) for two weeks, eventually mimicking adult values.
· a) Preterm infants: phase two can last more than one month.
· b) Exclusively breastfed infants: phase two can last more than one month.
· Pathological jaundice: In maternal serum jaundice (Lucey-Driscoll syndrome), jaundice occurs during the first 4 days of life. Characteristics: bilirubin is > 20 mg/dl; kernicterus is common; and resolves late.
· Typical causes for neonatal jaundice include normal physiologic jaundice, jaundice due to formula supplementation, and hemolytic disorders that include hereditary spherocytosis, G6PD deficiency, pyruvate kinase deficiency, ABO or Rh blood type autoantibodies, or infantile pyknocytosis. Serum bilirubin normally drops to a low level without any intervention required. In cases where bilirubin rises higher, a brain-damaging condition known as kernicterus can occur, leading to significant disability. A Bili light is used for early treatment, which often consists of exposing the baby to intensive phototherapy.
· Note: Two types of jaundice may occur in newborns who are breastfed. Both types are most often harmless.
· a) Breastfeeding jaundice is seen in breastfed babies during the first week of life. It is more likely to occur when babies do not nurse well, or the mother's milk is slow to come in.
· b) Breast milk jaundice may appear in some healthy, breastfed babies after day 7 of life. It is likely to peak during weeks 2 and 3 but may last at low levels for a month or more. The problem may be due to how substances in the breast milk affect the breakdown of bilirubin in the liver. Breast milk jaundice is different than breastfeeding jaundice. In breast milk jaundice, the bilirubin may increase to levels as high as 20 mg/dL, necessitating the need for phototherapy and the discontinuation of breastfeeding.
{kind=link}
{kind=link}
· Hyperbilirubinemia in the term newborn: neonatal hyperbilirubinemia is defined as a total serum bilirubin level above 5 mg per dL (86 μmol per L). Although up to 60% of term newborns have clinical jaundice in the first week of life, few have a significant underlying disease. However, hyperbilirubinemia in the newborn period can be associated with severe illnesses such as hemolytic disease, metabolic and endocrine disorders, anatomic abnormalities of the liver, and infections.
· Jaundice typically results from the deposition of unconjugated bilirubin pigment in the skin and mucous membranes. Depending on the underlying etiology, this condition may present throughout the neonatal period.
· Unconjugated hyperbilirubinemia is the most common form of jaundice encountered by family physicians.
· Causes of neonatal hyperbilirubinemia can be classified into three groups based on the mechanism of accumulation: bilirubin overproduction, decreased bilirubin conjugation, and impaired bilirubin excretion.
· Ι) Increased bilirubin load:
· a) Hemolytic causes: Characteristics: increased unconjugated bilirubin level, >6% reticulocytes, hemoglobin concentration of <13 g/dL (130 g/ L).
· i) Coombs' test positive: Rh factor incompatibility, ABO incompatibility, minor antigens incompatibility.
· ii) Coombs' test negative: red blood cell (RBC) membrane defects (spherocytosis, elliptocytosis), RBC enzyme defects (G6PD deficiency, pyruvate kinase deficiency), medications (e.g., sulfisoxazole acetyl with erythromycin ethylsuccinate, streptomycin, vitamin K), abnormal RBCs (hemoglobinopathies), sepsis.
· b) Non-hemolytic causes - Characteristics: increased unconjugated bilirubin level, normal percentage of reticulocytes.
· i) Extravascular sources: cephalohematoma, bruising, central nervous system (CNS) hemorrhage, swallowed blood.
· ii) Polycythemia: fetal-maternal transfusion, delayed cord clamping, twin-twin transfusion.
· iii) Exaggerated enterohepatic circulation: cystic fibrosis, ileal atresia, pyloric stenosis, Hirschsprung's disease, breast milk jaundice.
· II) Decreased bilirubin conjugation: Characteristics: increased unconjugated bilirubin level, normal percentage of reticulocytes. Causes: physiologic jaundice; Crigler-Najjar syndrome types 1 and 2; Gilbert syndrome; hypothyroidism; breast milk jaundice.
· III) Impaired bilirubin excretion: Characteristics: increased unconjugated and conjugated bilirubin level, negative Coombs' test, conjugated bilirubin level of >2 mg/dL (34 μmol/L) or >20% of total serum bilirubin level, conjugated bilirubin in urine.
· Causes:
· a) Biliary obstruction: biliary atresia, choledochal cyst, primary sclerosing cholangitis, gallstones, neoplasm, Dubin-Johnson syndrome, Rotor's syndrome.
· b) Infection: sepsis, urinary tract infection, syphilis, toxoplasmosis, tuberculosis, hepatitis, rubella, herpes.
· c) Metabolic disorder: alpha 1 antitrypsin deficiency, cystic fibrosis, galactosemia, glycogen storage disease, Gaucher's disease, hypothyroidism, Wilson's disease, Niemann-Pick disease.
· d) Chromosomal abnormality: Turner's syndrome, trisomy 18 and 21 syndromes.
· e) Medications: aspirin, acetaminophen, sulfa, alcohol, rifampin, erythromycin, corticosteroids, tetracycline, etc.
· Hyperbilirubinemia in the newborn – algorithm:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· http://img.medscape.com/pi/emed/ckb/pediatrics_cardiac/1331339-1331345-974786-974916.jpg (TSB: total serum bilirubin; TcB: transcutaneous bilirubin)
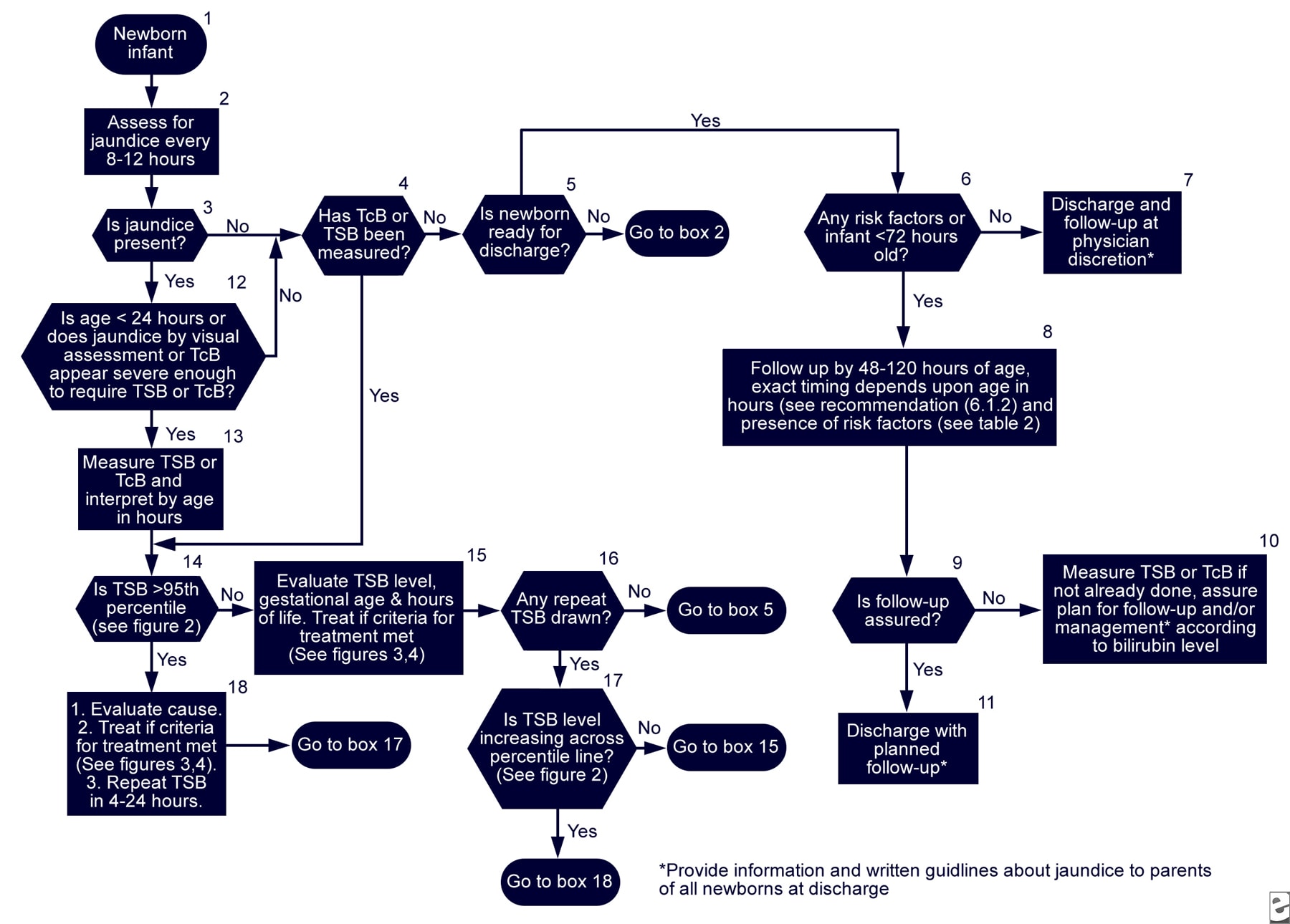{kind=link}
· Hyperbilirubinemia with abnormal liver function tests (LFTs) – diagnostic considerations: a focused history taking includes questions about recent medication use, travel, blood and body fluid exposure, alcohol consumption, and contaminated food.
· In acute viral or hepatotoxic injury, serum ALT, and AST are usually markedly elevated and can approach or exceed 1 000 U/L. Serum viral markers, including serologic tests for hepatitis A IgM, hepatitis B surface antigen, hepatitis B core IgM, and hepatitis C & E, should be evaluated routinely.
· Alcoholic hepatitis is often suggested by history. In alcoholic hepatitis, the serum AST (SGOT) /ALT (SGPT) ratio is often higher than 2:1, and the aminotransferase levels rarely exceed 300 U/L.
· An acute presentation of Wilson disease, an inherited disorder of copper metabolism, is very rare. Patients with Wilson’s disease demonstrate significant jaundice and modest elevations in AST, ALT, ALP, and GGT levels in conjunction with marked coagulopathy and other clinical features of a significantly decompensated liver disease. Coombs test-negative hemolytic anemia is usually present to some degree, the serum ceruloplasmin is low, and Kayser-Fleischer corneal rings may be apparent by ophthalmic slit-lamp examination.
· Autoimmune hepatitis, whether an acute episode or an acute relapse of chronic disease, may present with markedly elevated serum ALT and AST levels. An appropriate autoimmune screen includes antinuclear antibodies (ANAs), anti-smooth muscle antibodies (ASMAs), and protein electrophoresis to look for elevated gamma globulin levels.
· Cholestatic hepatobiliary disease: ALP (alkaline phosphatase) and γ-glutamyltransferase (GGT) are widely used as markers of cholestasis and are elevated disproportionately to the elevation of serum aminotransferases. The next step is abdominal ultrasonography to determine whether intrahepatic or extrahepatic biliary ducts are obstructed. The absence of biliary dilatation suggests intrahepatic cholestasis; the presence of biliary dilatation indicates extrahepatic cholestasis from mechanical obstruction.
· Intrahepatic cholestasis: causes of intrahepatic cholestasis can be categorized according to the pattern of bile duct injury and whether inflammation of the liver is acute or chronic. The diagnosis is often made by serologic markers in combination with liver biopsy.
· Infiltrative disease: the most common infiltrative liver diseases are granulomatous disorders, such as tuberculosis (TB) and sarcoidosis. Causes include Mycobacterium avium complex (MAC), fungal and parasitic infections, lymphoma, and toxins such as beryllium. Most of these conditions are accompanied by nonspecific systemic symptoms such as fever, night sweats, and weight loss. The presence of eosinophilia may suggest sarcoidosis, parasitic disease, or drug toxicity.
· Mechanical biliary obstruction: it may have a malignant or benign origin. Choledocholithiasis is found in about 15% of patients with gallbladder stones. The clinical presentation ranges from mild right upper quadrant pain with minimal elevation of liver enzymes to ascending cholangitis. Ultrasonography is as sensitive as CT for the detection of choledocholithiasis. ERCP is also highly accurate in the diagnosis of biliary obstruction, with a sensitivity of 89% to 98% and a specificity of 89% to 100%. ERCP can also be used for therapeutic interventions, including stone removal and endobiliary stent placement.
· Biliary obstruction may also be caused by parasitic infections such as with Ascaris lumbricoides. On endoscopy, the organisms can sometimes be seen protruding from the ampulla.
· Malignant causes include carcinoma of the pancreas or gallbladder, ampullary carcinoma, and cholangiocarcinoma. ERCP facilitates procedures such as diagnostic brushings or biopsy in cholangiocarcinoma and ampullary cancer, as well as the placement of an endobiliary stent to temporarily relieve the obstruction.
· Jaundice in primary sclerosing cholangitis (PSC) is typically a result of end-stage liver disease; however, occasionally a dominant nonmalignant extrahepatic biliary stricture is the cause, and this condition is treatable with endobiliary stent placement.
· Note: End-stage decompensated liver disease: it inevitably results in jaundice as the failing liver becomes unable to effectively excrete bilirubin. It may occur acutely in patients without previous liver disease, as in acute liver failure from drug toxicity (e.g., acetaminophen (paracetamol) overdose) or viral hepatitis. More commonly, it is seen in end-stage cirrhosis. Other indicators of decompensation include hepatic encephalopathy, ascites, renal impairment (such as hepatorenal syndrome), and (oesophageal) variceal hemorrhage. Laboratory findings include coagulopathy and, in chronic cirrhosis, hypoalbuminemia. The prognosis for an end-stage decompensated disease is usually poor and warrants consideration of liver transplantation.
· Child-Pugh score: it is used to assess the prognosis of chronic liver disease, mainly cirrhosis. It is used to determine the prognosis, as well as the necessary strength of treatment and the necessity of liver transplantation.
· It takes into account the following parameters: total bilirubin levels; serum albumin levels; prothrombin time (PT); the presence of ascites; and presence of hepatic encephalopathy.
· For the score & the interpretation see https://en.wikipedia.org/wiki/Child-Pugh_score
· Conjugated hyperbilirubinemia: It results from reduced secretion of conjugated bilirubin into the bile, such as occurs in patients with hepatitis, or from impaired flow of bile into the intestine, as in patients with biliary obstruction. Bile formation is sensitive to high levels of inflammatory cytokines such as in patients with septic shock.
· High levels of conjugated bilirubin may secondarily elevate the level of unconjugated bilirubin. Although the mechanism of this effect is not fully defined, one likely cause is reduced hepatic clearance of unconjugated bilirubin that results from the competition with conjugated bilirubin for uptake or excretion.
· Conjugated hyperbilirubinemia workup: all patients should be tested for:
· Full/ complete blood cell (FBC/ CBC) count e.g. to screen for hemolysis/ hemolytic anemia.
· Biochemistry: serum aminotransferases [aspartate aminotransferase (AST; SGOT), alanine aminotransferase (ALT; SGPT)]; alkaline phosphatase (ALP; if elevated or if an obstruction is suspected, images of the bile ducts, e.g., with ultrasonography should be obtained); Gamma-glutamyl transpeptidase (GGTP; may help differentiate a hepatic source of the elevated ALP from bone or other causes); fractionated bilirubin.
· Serology: serologic screen for viral hepatitis, including serology for EBV, CMV, HIV, hepatitis C virus (HCV) antibody and hepatitis B surface antigen (HBsAg) or anti-hepatitis B core antibody (anti-HBcAb); serological test for hepatitis A, D (HDV is considered to be a subviral satellite because it can propagate only in the presence of the hepatitis B virus) and E (HEV; common in developing countries e.g. in India);
· Toxicology: blood alcohol or acetaminophen (paracetamol; an analgesic/ antipyretic) levels upon admission (may be useful in some instances).
· Immunology: antimitochondrial antibody (to exclude primary biliary cirrhosis); antinuclear antibodies (ANAs), smooth-muscle antibodies, and other serologic studies (to exclude autoimmune hepatitis).
· Metals, alpha-1 antitrypsin & genetics: iron and genetic studies (when considering hemochromatosis); copper studies (to exclude Wilson disease); alpha-1 antitrypsin fractionation and other studies when considering hereditary liver diseases.
· Conjugated hyperbilirubinemia – causes Diagnostic considerations: CMV hepatitis, drug toxicity [such as acetaminophen (paracetamol), allopurinol, anabolic steroids, chlorpromazine, estrogens, halothane, isoniazid, methyldopa, phenytoin, protease inhibitors, quinidine, rifampicin, statins, and sulfa drugs], street drugs, environmental hepatotoxins (eg, beryllium), "nutraceuticals" such as herbal tea/ herbs (e.g. Kava, Black Cohosh, Boswellia Serrata and St John’s wort), organic solvents/ glue-sniffing; Amanita phalloides (mushrooms); acute fatty liver of pregnancy; inherited disorders of bilirubin conjugation [eg, Rotor syndrome (1*) & Dubin – Johnson syndrome (2*)]; liver congestion; liver ischemia (shock liver); rejection of transplanted liver; Reye syndrome (3*); total parenteral nutrition (TPN) toxicity and venous-occlusive disease associated with chemotherapy.
· Differential diagnosis: acute liver failure; acute pancreatitis; alcoholic hepatitis; amyloidosis; autoimmune hepatitis (4*); biliary obstruction; cardiac cirrhosis/ congestive (heart failure) hepatopathy; cholangiocarcinoma; cholangitis; cholecystitis; liver cirrhosis; gallstones (cholelithiasis); hemochromatosis; hemorrhagic shock; miliary tuberculosis (TB); pancreatic cancer; primary biliary cirrhosis (5*); sarcoidosis; septic shock; tricuspid regurgitation; viral hepatitis; Wilson disease (an autosomal recessive genetic disorder in which copper accumulates in tissues; it manifests as neurological/ psychiatric symptoms & liver disease (7*)); primary biliary cirrhosis.
· Intrahepatic causes of conjugated hyperbilirubinemia: hepatocellular disease [viral infections (hepatitis A, B, C, & E; Epstein-Barr virus (EBV) and cytomegalovirus (CMV)); chronic alcohol use; autoimmune disorders]; medications; pregnancy; parenteral nutrition; sarcoidosis; Dubin-Johnson syndrome; Rotor's syndrome; primary biliary cirrhosis; primary sclerosing cholangitis (6*).
· Extrahepatic causes of conjugated hyperbilirubinemia: intrinsic to the ductal system [gallstones; surgical strictures; infection (cytomegalovirus (CMV), Cryptosporidium infection in patients with acquired immunodeficiency syndrome); intrahepatic malignancy; cholangiocarcinoma]; extrinsic to the ductal system [extrahepatic malignancy (pancreas, lymphoma); pancreatitis].
· Note:
· I) Dose-dependent hepatotoxins include: some medications [acetaminophen (paracetamol; an analgesic & antipyretic), niacin (nicotinic acid) (used for hyperlipidemia), methotrexate (for immunosuppression, or chemotherapy))], ethanol, carbon tetrachloride (CCl4), vitamin A, metals (copper (Cu), iron (Fe), mercury (Hg)).
· II) Agents associated with idiosyncratic drug-induced liver disease (however drug-induced liver disease may be caused by any drug): isoniazid (used for tuberculosis), ketoconazole (an antifungal), amoxicillin & clavulanate, nitrofurantoin (the last 2 are antibiotics), dantrolene, estrogen, corticosteroids, diclofenac (an NSAID), sulindac, phenytoin (an antiepileptic), herbal preparations (e.g. kava; St Johns Wort; Black Cohosh, Boswellia Serrata).
· III) Drug-induced intrahepatic cholestasis: some medications may cause intrahepatic cholestasis or a mixed pattern of cholestasis and hepatitis. Drugs commonly associated with cholestasis include acetaminophen (paracetamol), penicillins, and anabolic and estrogenic corticosteroids. Cholestasis usually develops within 2 months of the initiation of therapy; it is generally reversible but may take many months to resolve. A biopsy may help establish a diagnosis of drug-induced intrahepatic cholestasis.
· (1*) Rotor syndrome is a rare, relatively benign autosomal recessive bilirubin disorder. It causes an increase in conjugated bilirubin. Rotor syndrome has many features in common with Dubin – Johnson syndrome (see below – differential diagnosis), an exception being that the liver cells are not pigmented. The primary symptom is non-itching jaundice. There is a rise in bilirubin in the patient's serum, mainly of the conjugated type. The SLCO1B1 and SLCO1B3 genes are involved in Rotor syndrome. Mutations in both genes are required for the condition to occur.
· (2*) Dubin – Johnson syndrome (DJS): is an autosomal recessive disorder that causes an increase of conjugated bilirubin in the serum without elevation of transaminases (ALT & AST). It is associated with a defect in the ability of hepatocytes to secrete conjugated bilirubin into the bile and is similar to Rotor syndrome. It is usually asymptomatic but may be diagnosed in early infancy based on laboratory tests. The conjugated hyperbilirubinemia is a result of the defective endogenous and exogenous transfer of anionic conjugates from hepatocytes (liver cells) into the bile. Impaired biliary excretion of bilirubin glucuronides is due to a mutation in the canalicular multiple drug-resistance protein 2 (MRP2).
· A darkly pigmented liver is due to polymerized epinephrine metabolites, not bilirubin. Unaffected subjects have a coproporphyrin III to coproporphyrin I ratio around 3 – 4:1, while in patients with DJS, this ratio is inverted, with coproporphyrin I being 3–4 times higher than coproporphyrin III. Analysis of urine porphyrins shows a normal level of coproporphyrin, but the I isomer accounts for 80% of the total (normally is 25%). High levels of GGT help in diagnosing pathologies involving biliary obstruction. Prognosis is good, and treatment of this syndrome is usually unnecessary. Most patients are asymptomatic and have normal lifespans. Some neonates present with cholestasis. It should be noted that hormonal contraceptives and pregnancy may lead to overt jaundice and icterus.
· Note: Differential diagnosis between Rotor & Dubin – Johnson syndrome: a) Appearance of the liver: On Rotor syndrome there is normal histology & appearance; on Dubin – Johnson syndrome liver has black pigmentation. b) Gallbladder visualization: On Rotor syndrome gallbladder can be visualized by oral cholecystogram; on Dubin – Johnson syndrome gallbladder cannot be visualized. c) Total urine coproporphyrin content: On Rotor syndrome it is high with < 70% being isomer 1; on Dubin – Johnson syndrome it is normal with > 80% being isomer 1 (normal urine contains more of isomer 3 than isomer 1).
· (3*) Reye’s syndrome is a potentially fatal syndrome that has numerous detrimental effects on many organs, especially the brain and liver. It may also cause hypoglycemia. The classic features are rash, vomiting, and liver damage. The exact cause is unknown and, while it has been associated with aspirin consumption by children with a viral illness. However, it may also occur in the absence of aspirin use. The disease causes fatty liver with minimal inflammation and cerebral edema (swelling of the brain). The liver may become slightly enlarged and firm, and there is a change in the appearance of the kidneys. Jaundice is not usually present.
· (4*) Autoimmune hepatitis: autoimmune hepatitis, whether an acute episode of an acute relapse of chronic disease, may present with markedly elevated serum ALT and AST levels. An appropriate autoimmune screen includes antinuclear antibodies (ANAs), anti-smooth muscle antibodies (ASMA), and protein electrophoresis to look for elevated gamma globulin levels.
· (5*) Primary biliary cirrhosis (PBC): a rare progressive liver disease, is most frequently seen in middle-aged women. The most common presenting symptoms are fatigue and pruritus. It is characterized by liver inflammation and loss of small intrahepatic bile ducts and ductules. Antimitochondrial antibody (AMA) testing is 95% sensitive and 98% specific for PBC. Marked hyperbilirubinemia indicates diminished chances of long-term survival.
· (6*) Primary sclerosing cholangitis (PSC): is characterized by fibrosing inflammation in the intrahepatic and extrahepatic bile ducts. Most patients are asymptomatic at the time of diagnosis. This diagnosis should be considered in patients with a history of inflammatory bowel disease (IBD; such as Crohn’s disease & ulcerative colitis (UC)) who have abnormal liver function tests, especially elevated ALP (alkaline phosphatase) levels. The diagnosis is made by ERCP or magnetic resonance cholangiopancreatography (MRCP).
· (7*) Wilson disease or hepatolenticular degeneration is an autosomal recessive genetic disorder in which copper (Cu) accumulates in tissues. It manifests as neurological or psychiatric symptoms and liver disease that may lead to liver failure and the need for liver transplantation. The condition is due to mutations in the Wilson disease protein (ATP7B) gene. Symptoms usually appear between the ages of 6 and 20 years, but cases in much older people have been described. Wilson's disease occurs in 1 to 4 per 100,000 people. Kayser – Fleischer rings are a pathognomic sign; these rings may be visible in the cornea of the eyes on slit-lamp examination as deposits of copper in a ring around the cornea. You may have a look at https://en.wikipedia.org/wiki/Wilson%27s_disease#/media/File:Kayser-Fleischer_ring.jpg
{kind=link}
· Unconjugated hyperbilirubinemia – causes:
· I) Hemolysis. Hemolytic disorders are either inherited or acquired. Inherited disorders, such as spherocytosis or sickle cell anemia, may be suggested by other laboratory abnormalities, including peripheral blood smear, elevated levels of lactate dehydrogenase (LDH), and decreased levels of haptoglobin. In these settings, serum bilirubin levels rarely exceed 5 mg/dL.
· II) Ineffective erythropoiesis (early labeled bilirubin (ELB) production): onset of asymptomatic jaundice. This is characterized by a marked increase in fecal urobilinogen excretion and a normal or near-normal red blood cell lifespan. A marked increase in ELB formation has been documented in diseases associated with ineffective erythropoiesis, including iron deficiency anemia; pernicious anemia; thalassemia; erythropoietic porphyria; lead poisoning; physiologic neonatal jaundice.
· III) Crigler-Najjar syndrome type 1: Crigler-Najjar type 1 is a scarce condition caused by the absence of bilirubin UDP glucuronyltransferase (UGT-1) activity. This syndrome usually develops during the neonatal period and leads to infant death. Apart from jaundice, the affected infant usually appears healthy at birth. Jaundice develops in the first few days of life and rapidly progresses by the second week; patients may present with evidence of kernicterus, the clinical manifestations of which are hypotonia, deafness, oculomotor palsy, lethargy, and, ultimately, death. Therefore, exchange transfusion is warranted despite phototherapy. Apart from jaundice, physical findings are usually normal in Crigler Najjar syndrome type 1, with no signs of hemolysis or liver disease. Except for the presence of high serum unconjugated bilirubin levels, the results of liver tests in Crigler-Najjar syndrome type 1 are normal. Serum bilirubin levels range from 20 – 50 mg/dL. Conjugated bilirubin is absent from serum, and bilirubin is not present in urine. A family history that includes consanguinity, relatives with severe jaundice without hemolysis, or relatives with evidence of liver disease and a history of exchange transfusion further supports the diagnosis. Because of its autosomal recessive transmission, consanguinity is a risk factor for Crigler-Najjar syndrome type 1. Definitive diagnosis of Crigler-Najjar syndrome requires high-performance liquid chromatography (HPLC) of bile or a tissue enzyme assay of a liver biopsy sample.
· IV) Crigler-Najjar syndrome type 2: is more common than type 1 and less severe. The bilirubin UDP glucuronyltransferase (UGT-1) activity is usually markedly reduced; however, phenobarbital increases this activity and reduces jaundice. Patients with this disorder have a normal life expectancy. Commonly, no clinical symptoms are reported with this disease. However, bilirubin encephalopathy has been reported. Patients with Crigler-Najjar syndrome type 2 appear healthy at birth, with no signs of liver disease. Patients may present with evidence of kernicterus. Crigler-Najjar syndrome type 2 results in lower bilirubin concentrations than does type I, with levels ranging from 7 – 20 mg/dL.
· V) Gilbert syndrome: the most common of the 3 inherited conditions, is found in 3% to 7% of the American population. The conjugation process is impaired because of reduced bilirubin UDP glucuronosyltransferase activity. This syndrome is benign and rarely produces clinical jaundice. Serum bilirubin levels may rise 2- or 3-fold with fasting or dehydration but are almost always less than 6 mg/dL. It may manifest only as jaundice on clinical examination; at least 30% of patients with Gilbert syndrome are asymptomatic, although nonspecific symptoms, such as abdominal cramps, fatigue, and malaise, are common.
· Abdominal symptoms may be multifactorial, with underlying anxiety probably playing an important role. Although it is true that not all patients with Gilbert syndrome and abdominal symptoms are anxious, they nonetheless appear to have organic-type discomfort that is hard to characterize and frequently eludes diagnosis.
· Gilbert syndrome can be diagnosed by a thorough history and physical examination and confirmed by standard blood tests. No relationship exists between abdominal symptoms and plasma bilirubin levels. Apart from mild jaundice, physical examination findings in people with Gilbert syndrome are normal.
· Lab results include unconjugated hyperbilirubinemia noted on several occasions; normal results from a full/complete blood count (FBC/CBC), reticulocyte count, and blood smear; normal liver function test (LFT) results; absence of other disease processes. Diagnosis is made in patients who have no past history of liver disease and manifest only jaundice on clinical examination. Specialized tests that have occasionally been used to confirm a diagnosis of Gilbert syndrome include fasting test; nicotinic acid test; phenobarbital test; radiolabeled chromium test; thin-layer chromatography; drug clearance test; polymerase chain reaction (PCR) assay; percutaneous liver biopsy (very rarely performed).
· Rarely, certain drugs may decrease bilirubin uptake. A prime example is rifampin, which may exacerbate unconjugated hyperbilirubinemia in those with Gilbert syndrome.
· VI) Physiologic neonatal jaundice: clinically evident in 50% of neonates during the first 5 days of life. In physiologic jaundice, the peak total serum bilirubin level is 5 – 6 mg/dL (86 – 103 µmol/L), occurs at 48 – 120 hours of age, and does not exceed 17 – 18 mg/dL (291 – 308 µmol/L).
· VII) Nonphysiologic neonatal jaundice: In maternal serum jaundice (Lucey-Driscoll syndrome), jaundice occurs during the first 4 days of life.
· VIII) Breast milk jaundice: bilirubin may increase to levels as high as 20 mg/dL, necessitating the need for phototherapy and the discontinuation of breastfeeding.
· Note: physiologic VS non-physiologic neonatal jaundice: While no specific test exists for physiologic jaundice, other causes of jaundice should be considered in infants with one or more of the following: jaundice occurring within 24 hours of birth; serum concentrations of unconjugated bilirubin of 11 – 12 mg/dL in infants who are formula-fed or 14 – 15 mg/dL in infants who are breastfed; increased levels of conjugated bilirubin (>2 mg/dL); jaundice persisting for more than 2 weeks.
· IX) Hematoma resorption.
· X) Other causes: Other conditions to be considered include: acute and chronic liver disease; primary hyperbilirubinemia from ineffective erythropoiesis; infections; cardiac disease (such as congestive heart failure or prosthetic heart valves); rhabdomyolysis; high-altitude living; medications (such as probenecid, rifampicin, or other antibiotics); thyrotoxicosis. Other differentials: dyserythropoiesis; portosystemic shunts; ethinyl estradiol.
· Differential diagnosis: iron-deficiency anemia; pediatric lead toxicity; pernicious anemia.
· Hemolysis/ hemolytic anemia (overview): hemolytic anemia occurs when the bone marrow is unable to replace the red blood cells that are being destroyed.
· Immune hemolytic anemia occurs when the immune system mistakenly sees the person’s own red blood cells as foreign substances. Antibodies then develop against the red blood cells (RBCs). These antibodies attack the red blood cells and cause them to break down too early.
· Causes: Red blood cells may be destroyed due to:
· a) Genetic defects within the red cells such as sickle cell anemia, thalassemia, and G6PD deficiency.
· b) Exposure to certain chemicals, drugs, and toxins.
· c) Infections.
· d) Blood clots in small blood vessels.
· e) Transfusion of blood from a donor with a blood type that does not match with the recipient.
· Workup: full/ complete blood count (FBC/ CBC) can help diagnose anemia and offer some hints to the type and cause of the problem. Essential parts of the CBC include red blood cell count (RBC), hemoglobin, hematocrit (HCT), and platelet (Plt) count.
· The following tests can identify the type of hemolytic anemia: absolute reticulocyte count; direct antiglobulin test (DAT; also known as direct Coombs test); indirect Coombs test; Donath – Landsteiner test (a test for antibodies related to paroxysmal cold hemoglobinuria); cold agglutinins (a test for cold agglutinin disease); free hemoglobin in the serum or urine; hemosiderin in the urine; serum protein electrophoresis; pyruvate kinase activity (to check for pyruvate kinase deficiency); osmotic fragility test (to exclude hereditary spherocytosis), serum haptoglobin levels; serum LDH; and urine and fecal urobilinogen.
· Features of hemolysis: in hemolysis unconjugated bilirubin is increased (while conjugated bilirubin is normal); urinary urobilinogen is increased; urine color is normal; stool color is normal; ALP (alkaline phosphatase) levels are normal; AST (SGOT) & ALT (SGPT) levels are normal; conjugated bilirubin in urine is not present; splenomegaly may be present.
· On the FBC/ CBC (full/ complete blood count) hemolytic anemia is indicated by decreased hemoglobin and/or hematocrit (a sign of anemia) and is also indicated by the presence of increased reticulocytes that also are present on the peripheral blood smear (polychromatophilic erythrocytes). On hemolysis, there are also elevated levels of (LDH) lactate dehydrogenase, and decreased levels of plasma haptoglobin.
· Direct antiglobulin test (DAT; also called direct Coombs test) is positive on autoimmune hemolytic anemia.
· Other features of hemolytic anemia include increased free serum hemoglobin, hemoglobin in the urine (hemoglobinuria), and hemosiderin present in the urine (hemosiderinuria).
· In autoimmune hemolytic anemia and hereditary spherocytosis, there is spherocytosis (presence of spherocytes) in the peripheral blood smear.
· Fractured red blood cells (schistocytes) in the blood smear are indicative of microangiopathic hemolytic anemia (MAHA) or other cause for intravascular hemolysis including disseminated intravascular coagulopathy (DIC)).
· Microangiopathic hemolytic anemia may occur in conditions such as hemolytic-uremic syndrome (HUS), thrombotic thrombocytopenic purpura (TTP; Moschcowitz syndrome), DIC (disseminated intravascular coagulation), pro/eclampsia, malignant hypertension; or as the RBCs (red blood cells) pass across a damaged or prosthetic heart valve. Other causes include severe burns, uremia, toxins, MDS (myelodysplastic syndrome), march hemoglobinuria, and hemolytic anemias caused by physical agents.
· Moreover, methemalbuminemia (the presence of methemalbumin in the circulating blood) is indicative of intravascular hemolysis.
· Note: methemalbumin is an albumin complex consisting of albumin and heme. This complex gives a brown color to plasma and occurs in hemolytic and hemorrhagic disorders. The Schumm test is used to differentiate intravascular hemolysis from extravascular hemolysis, as in hemolytic anemias. A positive result is indicative of intravascular hemolysis.
· Jaundice in the traveler to the tropics – differential diagnosis: obstructive jaundice due to biliary ascariasis; hepatic amebic abscess; acute viral hepatitis; malaria; enteric fever.
· a) Obstructive jaundice due to biliary ascariasis: it would give laboratory and imaging results suggestive of extrahepatic cholestasis: higher alkaline phosphatase (ALP) levels, lower serum aminotransferases, and dilated biliary tree. Differential: the above would also be the findings in a patient with choledocholithiasis or an obstructing pancreatic or ampullary lesion.
· b) Hepatic (liver) amebic abscess: is usually associated with mild or no abnormalities in the liver chemistries. Jaundice is uncommon, and the 100-fold elevation in the serum aminotransferase levels would never be observed. Additionally, ultrasound and MRI are sensitive tests for this diagnosis as well as for other focal liver diseases, such as pyogenic liver abscess and cancer.
· c) Malaria: causes jaundice as a consequence of hemolysis. Thus, there would be anemia, unconjugated hyperbilirubinemia, and an abnormal peripheral blood smear. Also, in malaria, liver injury is not severe enough to increase the aminotransferases more than 1- or 2-fold.
· d) Enteric fever: it is an essential disease in travelers; it is caused by Salmonella typhi (Salmonella enterica subsp. enterica serovar typhi)). The first 1 – 2 weeks of illness are similar to that found in our patient: generalized constitutional symptoms with fever, sweats, anorexia, and weight loss. However, by the third week of illness, there is extreme prostration and toxicity along with other physical findings, such as splenomegaly. This infection would have been suspected if the serum aminotransferase levels had not been so high – a 100-fold elevation in the aminotransferases is not in the clinical spectrum of typhoid fever. Differential: other systemic infections that can have prominent hepatitis include: Q fever, histoplasmosis, tuberculosis, brucellosis, syphilis, toxoplasmosis, leptospirosis, ehrlichiosis, and tularemia.
· e) Acute viral hepatitis.
· Note: the detailed history will assess for drug and toxins ingestion; also acute autoimmune hepatitis should be ruled out. If routine viral serologies for hepatitis A, B, and C, & D (HDV is considered to be a subviral satellite because it can propagate only in the presence of the hepatitis B virus) are negative, the diagnostic tests that would be considered include serology for EBV, CMV, HIV and Hepatitis E (assays for detecting anti-HEV IgM and IgG should be ordered). An infectious mononucleosis-like syndrome with prominent hepatitis can be seen with acute infection with cytomegalovirus (CMV), Epstein-Barr virus (EBV), and human immunodeficiency virus (HIV). A more common cause of acute hepatitis in India is hepatitis E virus (HEV) infection.
· HEV is the most common cause of epidemics of acute hepatitis in developing countries. Epidemic outbreaks periodically occur after periods of more substantial than usual rainfall. However, HEV is also a significant cause of sporadic hepatitis in endemic areas. HEV infection accounts for more than 50% of cases of acute viral hepatitis in young adults in developing countries.
· In industrialized countries, cases of acute HEV infection are reported but are uncommon. In developed countries, the disease is primarily – but not exclusively – recognized in travelers returning from endemic areas or in individuals consuming undercooked pig or deer meat. The seroprevalence of HEV antibodies in US blood donors is 1% to 2%, but in some locations is as high as 20%.
· Anti-HEV IgM becomes detectable just before the peak ALT activity and declines weeks to months after the ALT begins to normalize. Anti-HEV IgG becomes detectable within days of the production of IgM antibodies, increases throughout the icteric phase, and has been detected up to 14 years after acute infection. Many patients with acute HEV infection have both IgM and IgG antibodies. Qualitative assays of hepatitis E RNA from serum, liver, or stool using reverse transcriptase-polymerase chain reaction (RT-PCR) exist but are used primarily for research purposes. Fulminant hepatitis occurs in 1% to 3% of nonpregnant patients with acute HEV infection and in as many as 25% of pregnant women with acute HEV infection.
· The Indian epidemic in Myanmar in 1977 was associated with an 18% mortality rate among pregnant women. Maternal fatality increases with an increased term at the time of infection. In the Chinese epidemic in the 1980s, fatality rates in the first, second, and third trimesters were 1.5%, 8.5%, and 21%, respectively. Superinfection with HEV in patients with underlying chronic liver disease can cause severe liver decompensation that is frequently complicated by hepatic encephalopathy and renal failure and has a protracted course with high morbidity and mortality.
· Note: Liver biopsy is not routinely performed, but when there is diagnostic uncertainty, a suspicion of multiple diagnoses, or suspicion of acute-on-chronic liver disease with progressive deterioration, a biopsy may be helpful. In a female with acute hepatitis, autoimmune hepatitis must be ruled out by searching the biopsy for plasma cells.
No comments:
Post a Comment